







提示:文章写完后,目录可以自动生成,如何生成可参考右边的帮助文档
文章目录
前言
提示:这里可以添加本文要记录的大概内容:
蛋白质是生命活动的重要执行者,其功能与结构密切相关。蛋白质结构从一级到四级,层层递进,决定了其生物学功能。随着计算生物学的发展,蛋白质三级结构的预测、可视化与分析成为研究蛋白质功能的重要手段。本文将从蛋白质结构的基本概念出发,介绍三级结构的预测方法、可视化工具以及相关计算技术,为蛋白质结构与功能研究提供参考。
一、蛋白质的结构
1.1 蛋白质的结构
蛋白质的结构分为四级:
一级结构Primary structure 氨基酸序列
二级结构 Secondary structure 周期性的结构构象
三级结构 Tertiary structure 整条多肽链的三维空间四级结构级结构 Quaternary structure 多个亚基形成的复合体结构
1.2 蛋白质的二级结构
蛋白质经过折叠后会形成规则的片段,这些规则的片段构成了蛋白质的二级结构单元。三种常见的二级结构单元包括螺旋、β折叠、和转角。
螺旋中最常见的就是α螺旋,有其他的螺旋,比如 3 转角螺旋,5 转角螺旋等。
β折叠由平行排列的β折片组成。这些折片在序列上可能相隔很远,但是在空间结构上并排在一起,彼此间形成氢键。
除了螺旋和折叠外,蛋白质结构中还存在大量的无规律松散结构 coil。如果这些无规律的肽链突然发生了急转弯,这个转弯结构就叫做β转角。
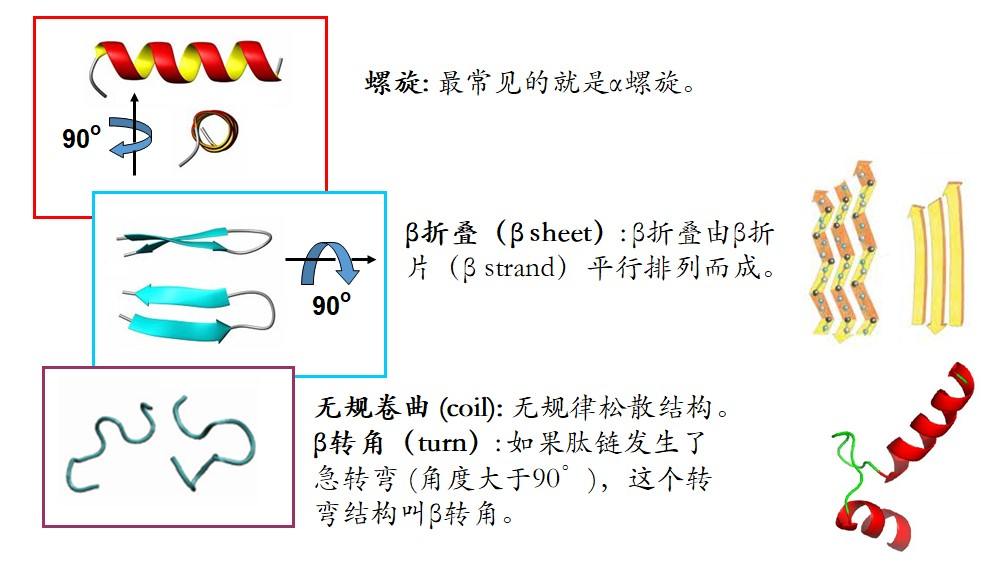
蛋白质的二级结构经常用图形来形象的描述。比如图中黄色的箭头代表对应的氨基酸具有β折片结构。波浪线代表螺旋结构,小鼓包是转角。此外,以字母形式书写的二级结构序列能够更加精准的描述。其中,E 代表β折叠,H 代表α螺旋,T 代表转角。没有写任何字母的地方是松散的 coil 结构。
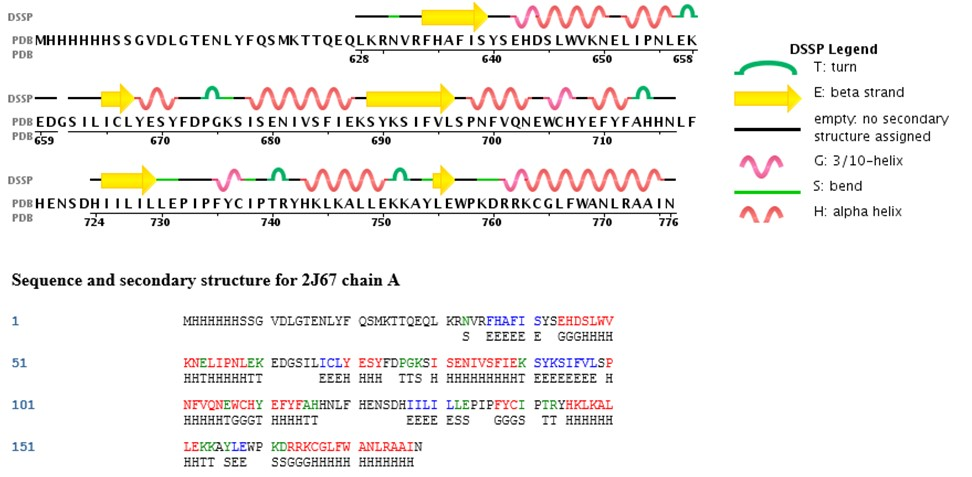
已知蛋白质二级结构获得
DSSP,也就是蛋白质二级结构定义词典。https://swift.cmbi.umcn.nl/gv/dssp/
研究人员根据 DSSP,将三级结构里的二级结构单元指认出来的。然后再按照规定的格式,记录下蛋白质中每个氨基酸处于哪种二级结构单元。这样一个记录蛋白质二级结构信息的文件叫做 DSSP 文件。蛋白质结构数据库 PDB 中的每一个蛋白质三级结构都有自己对应的 DSSP 文件。DSSP 文件里不同字母所代表的不同二级结构单元和 PDB 里面的记录方式是统一的。
PDB 数据库中有一个叫做“ss.txt”的文件:http://www.rcsb.org/pdb/files/ss.txt.gz (压缩文件 30.6M)。这个文件里面有 PDB 所有蛋白质结构的一级和二级结构的 FASTA 格式序列。但是这个文件非常大!仅仅打开文件就要耗费许久时间,使用起来相当的不方便。
页可以用老师编写的小程序http://1.51.215.28/~gongj/biotools/。只需要输入PDB ID,程序就会自动下载相应的 DSSP 文件,并从中抽取出一级和二级结构的序列信息,最后以 FASTA 格式输出。
未知蛋白质二级结构获得
要用计算机软件来预测蛋白质的二级结构。预测的结果和真实情况会有一定出入,究竟差多少,取决于预测软件的准确度。可以预测蛋白质二级结构的软件很多,而且都可以在线使用。

以 PSIPRED 为例,看看如何从氨基酸序列预测二级结构(图 1)。PSIPRED(http://bioinf.cs.ucl.ac.uk/psipred)是一个蛋白质序列分析平台,它不仅可以预测二级结构,还有很多其他分析功能,比如预测三级结构。选择第一个“PSIPRED”工具,即,预测蛋白质二级结构的工具。
1.3 蛋白质的三级结构
蛋白质的三级结构是指整条多肽链的三维空间结构,也就是包括碳骨架和侧链在内的所有原子的空间排列。
蛋白质的三级结构测定方法:X-射线衍射法、核磁共振法、冷冻电子显微镜技术。
获取蛋白质三级结构最直接的办法就是去 PDB 搜索(http://www.rcsb.org/)。
PDB 文件是通过记录蛋白质中每一个氨基酸上的每一个原子的三维坐标来存储空间结构信息的。这些原子坐标可以被三维可视化软件读取。三维可视化软件能够创建一个三维空间,然后根据原子坐标以及原子的大小把原子展示在空间内,并根据原子间的距离给它们连上化学键。这样一个立体的蛋白质结构就呈现在眼前了。
二、三级结构可视化软件VMD
能够实现蛋白质三维结构可视化的软件非常多。比 如 ,专业 级的PyMOL(https://pymol.org/2/)。这个软件已经被世界上著名的生物医药软件公司“薛定谔公司(Schrödinger)”收购。这种专业级的可视化软件不仅能够做出非常漂亮的图片,它还有强大的插件支持各种各样的蛋白质结构分析。
功 能 同 样 强 大 的 免 费蛋白质三维结构可 视 化软 件, VMD (http://www.ks.uiuc.edu/Research/vmd)。VMD 由伊利诺伊大学研发。下载 VMD 需要先注册获得一个账户,之后就可以根据你的操作系统和机器配置选择合适的版本下载了。
file & mouse
representation
multiple representations
display & lable
三、计算方法预测三级结构
3.1 介绍
通过 VMD 等三维可视软件可以看到了蛋白质的立体三维结构。可以通过诸如 X 射线衍射法、核磁共振法、以及冷冻电子显微镜技术等实验方法获得。但是这些实验方法普遍存在三个问题:(1)实验材料要求高,(2)实验仪器造价高,(3)实验耗时成本高。这也是为什么几百万个已发现的蛋白质序列中只有十万多个解析出三维结构的原因。
想要了解剩下那些蛋白质的结构,我们可以尝试利用计算机来预测。预测出来的结构模型虽然不是蛋白质的真实结构,但是如果蛋白质本身基础好的话,预测模型会十分接近它的真实结构。
预测蛋白质的三级结构,常用的方法有从头计算法(ab initio),同源建模法(homolog modeling),穿线法(threading)和综合法(ensemble method)。
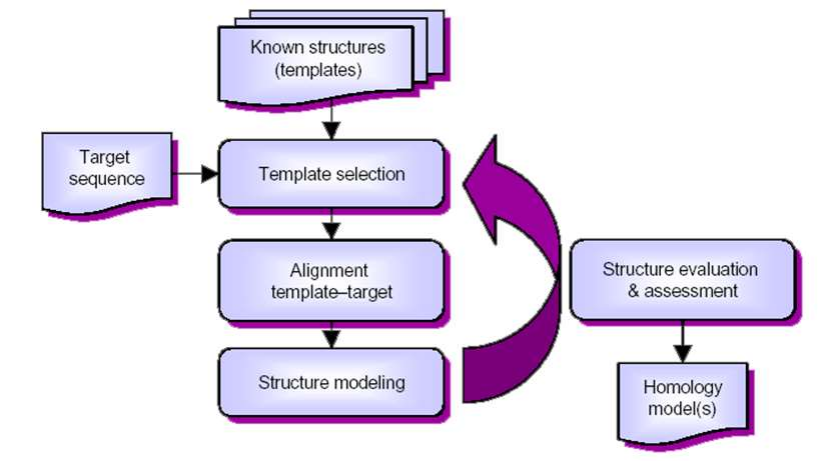
3.2 同源建模法 SWISS-MODEL
原理:相似的氨基酸序列对应着相似的蛋白质结构。
所以同源建模法的关键就是找到一个好的模板。好的模板要求,在序列水平上模板(template)要与目标(target)蛋白质具有超过 30%的一致度。
同源建模法操作流程如下:
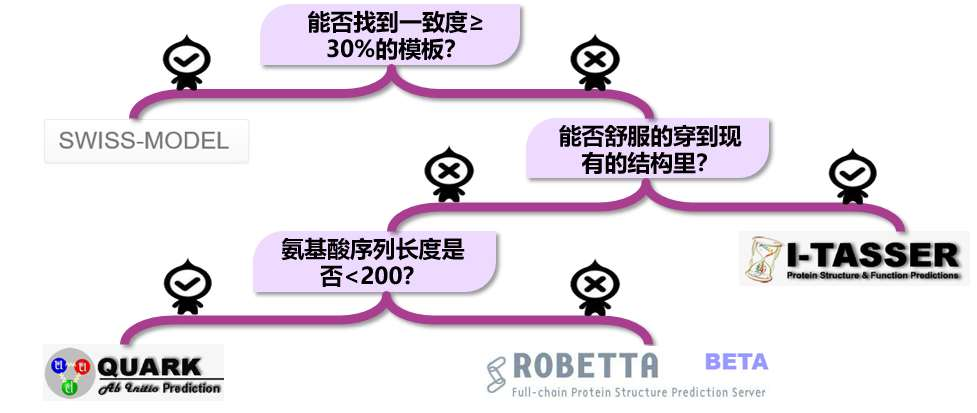
- 确定模板:找到与目标蛋白质同源的已知蛋白质结构作为模版(目标序列与模版序列间的一致度要≥30%)。
- 序列比对:为目标序列与模板序列创建序列对比。模板可以选取多个,通过做多序列比对,各取所长,让模板序列中与目标序列相似的片段尽可能多的覆盖整个目标序列,同时要尽量避免没有模板参考的断口。
- 计算模型:通过序列比对,将目标序列里的氨基酸替换到模板结构里对应的氨基酸所在的空间位置上。这一步通过同源建模软件来实现。
- 质量评估:同源建模软件输出结构模型后还需要进行质量评估,并根据评估结果更换模板或修正序列比对,重新构建模型,再次评估。重复这个过程,直至模型质量合格为止
SWISS-MODEL(http://swissmodel.expasy.org)是一款常用的全自动同源建模在线软件。它能帮助完成上述步骤中从模板选取到创建序列比对,再到计算模型,以及最后的质量评估的全部过程。你需要做的只是:输入目标序列,点 Build Model(创建模型)。大约三到五分钟之后就会返回结果。
预测效果:
如果目标序列与模板序列一致度极高,那么同源建模法是最准确的方法(图 4)。如果一致度如果达到 30%,那么模型的准确度就可以达到 80%,模型可以用于寻找功能位点以及推测功能关系等。如果一致度达到 50%,模型准确度就可以达到 95%,可以根据模型设计定点突变实验,甚至可以做晶体结构置换,也就是辅助完成真实结构的测定。如果一致度达到70%以上的话,我们基本就可以认为预测模型完全代表了真实结构,并可以用它进行虚拟筛选、分子对接、药物设计等结构功能研究。但是,“序列越相似,结构越相似”也有例外情况。虽然同源建模法是蛋白质三级结构预测的首选方法,但是对于那些找不到“好”模板的蛋白质,此方法并不适用。
3.3 穿线法 I-TASSER
虽然同源建模是蛋白质结构预测的首选方法,但是对于那些找不到合适模板(一致度大于 30%的模板)的蛋白质,此方法并不适用。这种情况下可以尝试穿线法(threading)。穿线法基于的原理是:不相似的氨基酸序列也可以对应着相似的蛋白质结构。
PDB 数据库里已解析出的真实蛋白质结构每天都会增加,但是从 2008 年开始就再没有新的结构拓扑产生了。换言之,目前发现的蛋白质结构的种类已定格在 2008 年的 1393种了,即已知的 10 万多个结构,根据结构拓扑划分,分成了 1393 种。新解析出来的结构必定属于其中之一。而具有同一结构拓扑的蛋白质,序列水平上有相似的,也有不相似的。

穿线法就是通过计算目标序列穿到每一个已知结构中的每一种穿法下的能量,找到能量最低的那种穿法以及所穿的结构,然后把目标序列中的氨基酸替换到模板结构里来构建结构模型的。
用穿线法预测蛋白质三级结构,首选密歇根大学张阳教授的 I-TASSER(http://zhanglab. ccmb.med.umich.edu/I-TASSER)。这个预测系统在 CASP 第 7 到第 11 届蛋白质结构预测比赛中都名列第一。CASP 全称是蛋白质结构生物信息预测国际竞赛。两年一届。每次比赛,参与者们会对一组即将公开的结构进行预测。再将预测模型和真实结构进行比较,看谁预测的最准。
张阳教授的 I-TASSER 可以在线提交预测任务,不需要提前下载安装。提交氨基酸序列进行预测前需注册获得用户名密码,注册是完全免费的。再给任务起个名字。最后点Run I-TASSER。
3.4 从头计算法 QUARK
用同源建模法解决不了的,一般用穿线法就可以解决。但是也有一些特殊的蛋白,穿线法也解决不了。这种蛋白质,尽管可以用 ITASSER 做出模型,但如果看一下模型的质量评
估系数,都不合格。这时我们就得采取其他方法。可以尝试从头计算法(ab initio)。从头计算法的原理基于 1973 年 Anfinsen 在Science 上提出的一句话。他说,蛋白质
的三维结构决定于自身的氨基酸序列,并且处于最低自由能状态。
也就说,当一个蛋白质被翻译出来之后,它上面的氨基酸排列顺序就已经决定了这个蛋白质长什么样子。一个蛋白子所应该具有的天然模样就是它的氨基酸序列所能摆出的自由能最低的 pose,因为能量低意味着稳定和持久。所以从头计算法会模拟肽段在三维空间内所有可能的存在姿态,并计算每一个姿态下的自由能,最终给出自由能最低的那个姿态作为预测结果。可想而知,这是多么大的计算量啊!
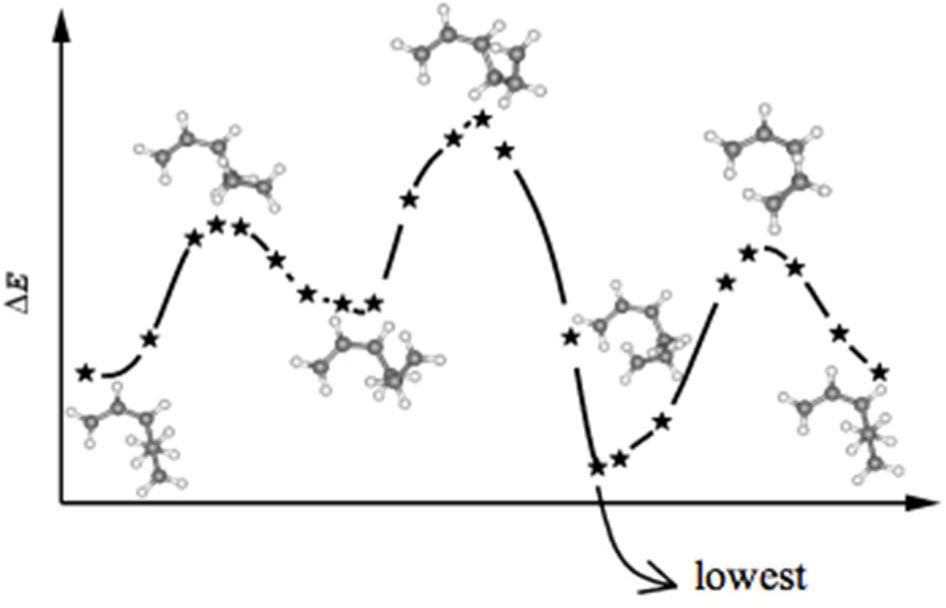
做从头计算法的预测软件不多,首选张扬教授的 QUARK 预测软件(http://zhanglab.ccmb. med.umich.edu/QUARK)。注意,QUARK 只能为长度在 200 个氨基酸以内的蛋白质预测结构。输入序列和用户信息。QUARK 需要单独注册,ITASSER 的账号在这里不通用。QUARK需要很长的计算时间,大约需要 2 天以上才能计算出结果。
3.5 综合法 ROBETTA
除了同源建模法、穿线法和从头计算法,还有第四种预测方法,综合法(ensemble method)。综合法综合了前三种方法,将氨基酸序列分段,情况不同的片段采用不同的方法预测。
ROBETTA 是一款使用综合预测三级结构的软件,它综合了同源建模法和从头计算法两种方法。能找到模板的区域用同源建模法,找不到的区域用从头计算法。ROBETTA 网站同样需要注册(http://robetta.bakerlab.org)。
ROBETTA 需要的等待的时间是四种方法里最长的,首先任务要排队,至少要排两个小时。排队排上之后,还要等几个小时,才会出现分析页面。分析页面里,输入的氨基酸序列被分成了几段,比如前两段适合用从头计算法预测,后两段适合用同源建模法预测。之后,点击分段链接,逐段进行预测。每一段的预测时间都不短。同源建模法预测的,需要几个小时到几天的时间,从头计算法预测的,需要几天到几周的时间,这取决于目标序列的预测“难易”程度。

3.6 总结
什么情况下用什么软件。要预测一个蛋白质的三级结构,首先看能不能找到一致度大于等于 30%的模板,如果能,用 Swiss-model,又快又准。如果不能,再看能否用 I-TASSER 预测出合格的模型。如果不能,再看氨基酸序列的长度是否小于 200,如果小于 200,用 QUARK。如果大于 200,用 ROBETTA!如果,ROBETTA也做不出质量合格的模型,那说明蛋白质太特殊了,现有的软件都不适用。这种情况你需要联系专门的研发小组,为你的蛋白开发专门的软件。
3.7 模型质量评估
软件预测出结构模型后还需要对模型进行质量评估,以确定模型是否可靠。可靠的模型才能发表或进行下一步研究,不可靠的模型一点用处都没有。模型质量评估软件并不比较预
测模型跟真实结构之间的差别大小,而是从空间几何学、立体化学和能量分布三方面评估一个模型的自身合理性。
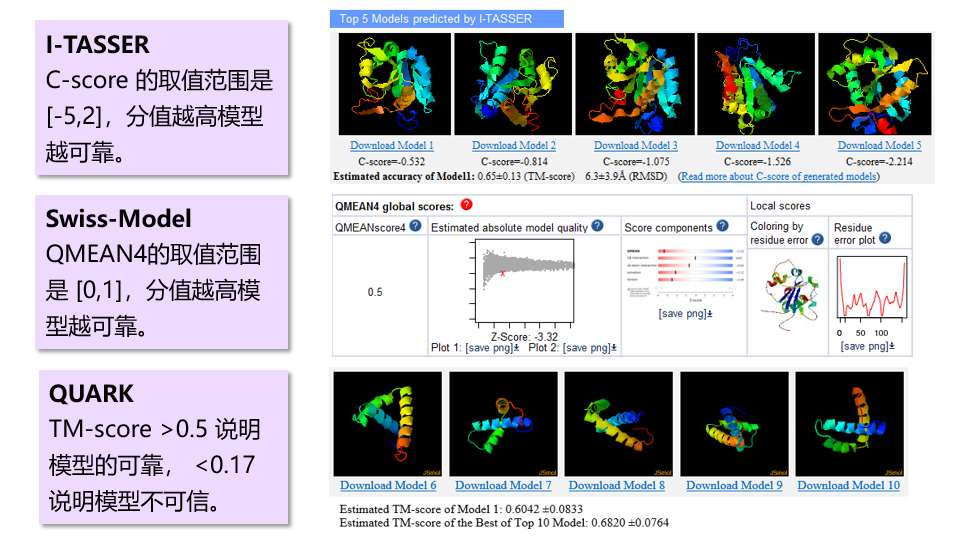
大多数预测软件都自带模型质量评估系数,比如 I-TASSER 有 C-score,Swiss-Model 有QMEAN score,QUARK 有 TM-score 等(图 1)。这些系数可以一定程度上反应模型的质量,但不能完全反应。至少要有三个评估体系都认为模型可靠,模型才真的可靠。这就需要借助第三方的评估软件。
SAVES 是常用的第三方的评估软件之一(http://services.mbi.ucla.edu/SAVES/)。SAVES平台提供了 6 种评估软件。可上传一次模型的 PDB 文件,然后 6 个软件同时评估。也可以单独运行最常用的 Verify3D 和 PROCHECK 评估软件。
Verify3D 会根据模型质量,返回一个 3D-1Dscore。只有超过 80%的残基拥有大于 0.2 的 3D/1D score,才能被认为是质量合格的模型。从 3D-1Dscore 分布图上可以查看,模型中哪些位置分值比较低,然后针对这些低质量区域进一步修正。
PROCHECK 可以做出模型结构的拉氏图。拉氏图检查 Cα的两面角是否合理。合格的模型超过 90%的残基都应该位于红色的允许区域和正黄色的额外允许区域内。落到其他区域的残基应当被查看并修正。
此外,ProQ 也是一个快速好用的评估软件(http://www.sbc.su.se/~bjornw/ProQ)。它的评估结果十分明确,LGscore 和 MaxSub 两个值所处范围直接对应模型的好坏。
ModFOLD 通过 P-value,给出模型的可信度(http://www.reading.ac.uk/bioinf/ModFOLD)。但是 ModFOLD 较其他几种方法,评估需要较长的时间。其他的都是立等可取,ModFold 需要半个小时。而且一个 email 地址一次只能提交一个任务。
四、三级结构的比对
蛋白质三级结构的比对不同于序列比对,它是针对蛋白质三维空间结构的相似性进行比较,是蛋白质结构分析的重要手段之一。
结构比对可用于探索蛋白质进化及同源关系、改进序列比对的精度、改进蛋白质结构预测工具、为蛋白质结构分类提供依据,以及帮助了解蛋白质功能等。
结构比对的结果可以用很多种参数来衡量,最常用的是 root mean squared deviations (RMSD)。如果两个结构的 RMSD 为 0 埃,那么就认为它们结构一致,可以完全重合。一般来说,RMSD 小于 3 埃时,认为两个结构相似。
4.1 SuperPose 叠合
SuperPose 是一款在线蛋白质结构叠合软件(http://wishart.biology.ualberta.ca/SuperPose/),可以将两个结构比对在一起,并给出两者之间的 RMSD。
4.2 SPDBV 选择叠合
PDBV 是一款拥有强大结构分析功能的绿色软件,无需安装,下载后直接运行即可(http://spdbv.vital-it.ch/)。SDPBV 有分子查看器的功能,也是一个蛋白质同源建模
平台,同时它还能为两个结构进行整体智能叠合,或者进行选择性叠合。
如果我们想要把相同的结构分毫不差的叠合起来,以看清楚不同区域的差别的话,就需要用到选择性叠合。
五、蛋白质分子表面性质
蛋白质分子表面性质是蛋白质结构的重要研究内容之一,对了解蛋白质的功能至关重要。
蛋白质的表面性质包括表面性状、表面电荷分布、表面残基可溶性等。
表面性状可以用 VMD的 Surface 这种 Representation 表示,电荷分布和可溶性分布同样也可以用 VMD 计算并标识出来。
5.1 VMD 创建 PSF 文件
VMD 做蛋白质表面的电荷分布(课程附件下载 A.pdb)
- 下载 APBS 插件
做电荷分布,需要先给 VMD 下载 APBS 的插件(可从课程附件中下载),下载后解压缩得到 APBS 文件夹。将 APBS 文件夹移动到 VMD 的安装目录里(也可放在其他位置)。需要说明的是,VMD 不识别中文,所以不管是读取的文件还是写入的文件,你的文件路径中都不能有中文。 - 创建工作目录
首先在 D 盘的根目录下创建一个工作文件夹 test。之后产生的一系列文件都存在这里。把 A.pdb 移动到 test 文件夹里。 - 创建 PSF 文件
PSF 文件是另一种存储蛋白质结构信息的文件。做表面电荷分布的 APBS 插件需要读取通过 A.pdb 创建出的 PSF 文件。
- 打开 VMD,读取 A.pdb
- 点主窗口 Extensions → 点 Modeling → 点 Automatic PSF Builder → 弹出 AutoPSF 窗口
- 在 AutoPSF 窗口的 Molecule 选择已打开的 A.pdb
- 在 Output basenam 里给即将创建的 PSF 文件起名并指定其保存路径为 D:\test\A_autopsf
- 在 Topology files 里面选中 VMD 自带的拓扑文件 → 点 Load input files 加载文件
- 在 Step2: Selections to include in PSF/PDB 里选 everything → 点 Guess and split chains ……
- 弹出提示窗口 → 点 no
- 在 Step3: Segments Identified 里检测到一条链
- 点右下角 I’m feeling lucky,开始创建 PSF 文件
- 弹出提示窗口 → 点 no
- 弹出提示窗口 → 显示 structure complete → 点确定,PSF 文件创建成功
PSF 文件创建成功后,test 文件夹里多了三个文件:A_autopsf.log,A_autopsf.psf 和
A_autopsf.pdb 文件。Log 文件为日志文件,无用。下一步做蛋白质表面的电荷分布需要用到
另外两个新创建的 PDB 文件和 PSF 文件
5.2 APBS 计算表面电荷分布
六、获取蛋白质四级结构
蛋白质四级结构是独立的三级结构单元聚集形成的复合物,其中每个独立三级结构称为亚基,也称为单体(monomer)。含两个亚基的蛋白质称为二聚体(dimer),比如图 1 中 Toll样受体 3 的两个单体聚集在一起形成二聚体识别病原双链 RNA 的结构。再有比如血红蛋白的四聚体结构(tetramer)和热休克蛋白的六聚体结构(hexamer)。

在研究蛋白质结构功能时,可通过实验方法获得它们的四级结构。实验方法主要是采用 X 射线衍射法。这种方法可以获得复合体的真实结构,但是技术难度较大,主要是因为复合体很难获取并成功结晶。此外还有冷冻电子显微镜技术,但这种方法不能像 X射线衍射法像那样获得精准的真实结构。它只能捕获类似影子的外形轮廓,之后,再根据已有的同源蛋白质晶体结构对影子中的单体进行同源建模,再把模型嵌入到影子里,以此建出复合体的模型。
目前通过试验方法获得的四级结构都可以从 PDB 数据库中找到。此外,还有专门的蛋白质相互作用关系数据库。比如,DIP 数据库,专门用于存储实验方法测定的蛋白质之间的相互作用。BioGRID 数据库主要收集模式生物物种中涉及的蛋白质间相互作用。STRING 数据库除了实验方法测定的蛋白质间相互作用外,还储存已发表的用计算方法预测的蛋白质间相互作用。

七、蛋白质-蛋白质分子对接
除了实验方法和从数据库中获取,还可以通过计算机预测蛋白质的四级结构。这种预测技术叫分子对接(molecule docking)。
目前有很多做分子对接的软件,这些软件的基本思路都是尝试分子间所有可能的结合方式,并根据结合后能量的高低给每种结合方式打分、排名。分子结合后,能量越低说明这种结合方式越稳定。
软件在对接的过程中会考虑以下因素:
1)形状互补,
2)亲疏水性,
3)表面电荷分布。
分子对接分为蛋白质和蛋白质之间的分子对接以及蛋白质和小分子之间的分子对接。蛋
白质和蛋白质之间的分子对接又分为两种不同的方法,即刚性对接(Rigid Docking)和柔性对接(Flexible Docking)。
刚性对接是指对接过程中蛋白质是硬的,外形不可变,柔性对接
则反之。刚性对接实际上是一种近似计算。因为蛋白质在机体环境中是软的,他的表面是在微小移动中保持着动态平衡。但是要在对接过程中考虑到这个实际情况则需要庞大的计算量,所以能够模拟蛋白质真实状态的柔性对接软件非常少,且都是收费的软件。目前可用的大多数免费软件都是刚性对接软件。
蛋白质和蛋白质之间的刚性对接软件常用的有 ZDOCK(http://zdock.umassmed.edu/)和 GRAMM-X(http://vakser.bioinformatics.ku.edu/resources/gramm/grammx),这两个都是免费的刚性对接软件。HadDock(http://haddock.chem.uu.nl/)可以做蛋白质局部柔性对接,但需要和开发者联系以获取免费的使用权限。这些软件的输出值都是根据能量高低排序的多个对接模型,能量低者排名靠前。至于选取哪个模型作为最终结果需要综合考虑各种因素,并非一定选取排名第一的模型。
用 ZDOCK 软件预测出两个蛋白质最有可能的结合方式之后,还需要对预测的对接结果进行进一步分析。PDBePISA(http://www.ebi.ac.uk/pdbe/pisa/)能够在线分析蛋白质复合
体中的相互作用面。
八、蛋白质-小分子分子对接
蛋白质和小分子化合物之间的对接要比蛋白质和蛋白质之间的对接复杂一些。从体积上看,小分子要远远小于蛋白质,所以蛋白质和小分子之间的对接更像是钥匙与锁的关系,只有正确的钥匙以正确的姿态插入锁芯才能打开锁。蛋白质和小分子之间的对接也可分为刚性对接和柔性对接。值得注意的是,在这两种对接中,小分子总是柔性的,所谓的刚性柔性指的仍然是蛋白质分子。
以 Autodock(http://autodock.scripps.edu)这款免费对接软件为例,来演示蛋白质和小分子对接的过程。
Autodock安装
Autodock使用
九、虚拟筛选与反向对接
蛋白质与小分子对接方法:虚拟筛选
虚拟筛选,也称计算机筛选,即在进行性物活性筛选之前,在计算机上对化合物分子进行预筛选,以降低实际筛选化合物的数目,同时提高先导化合物的发现效率。
虚拟筛选:化合物小分子数据库ZINC http://zinc.docking.org
蛋白质与小分子对接方法:反向对接
反向对接(Target Fishing)是通过把一个小分子与多个靶标蛋白进行分子对接,寻找潜在的靶标。
当前软件和技术:还没有标准和成熟的免费软件来实现,只有少数收费软件能实现此功能,以及很少的科研单位通过自己的算法和对已有对接程序的改造来实现。
靶标数据库:http://bioinfo-pharma.u-strasbg.fr/scPDB
十、分子动力学模拟
分子动力学模拟(Molecular Dynamic Simulation,MDS)
一用计算机来模拟原子及分子的物理运动过程。
软件:NAMD,CHARMM,DESMOND,GAUSS,等
总结
蛋白质结构的研究是理解其功能的基础。通过同源建模、穿线法、从头计算等预测方法,结合VMD、APBS等工具,可以深入分析蛋白质的三级结构、表面性质及分子间相互作用。分子动力学模拟和分子对接技术进一步拓展了蛋白质研究的深度与应用范围。这些方法为蛋白质功能解析、药物设计等领域提供了强大的技术支持,推动了生命科学的发展。



















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











