






![]()
================================
分享一个 Warren Cowley Parameters 程序:表征短程有序的化学基序。
感谢论文的原作者!
================================
主要内容
“晶体材料的化学成分具有原子尺度的波动,可调节各种中尺度特性。建立此类材料的化学-微结构关系需要对这些化学波动进行适当的表征。然而,目前的表征方法(如 Warren-Cowley 参数)只能部分利用局部化学图案所包含的完整化学和结构信息。在此,我们介绍一种基于 E(3)- 等变图神经网络的框架,它能够完全识别任意化学元素数量的任意晶体结构中的化学图案。这种方法自然而然地为量化化学复杂材料中的化学短程有序(SRO)提供了一种适当的信息论度量,并为化学空间提供了一种精简但完整的表征。我们的框架可以将任何原子间性质与其相应的局部化学主题相关联,从而为探索化学复杂材料的结构-性质关系提供了新的途径。我们使用 MoTaNbTi 高熵合金作为测试系统,通过评估与每个化学主题相关的晶格应变,以及计算化学波动长度尺度的温度依赖性,展示了这种方法的多功能性。”——取自文章摘要。
================================
Figure 1
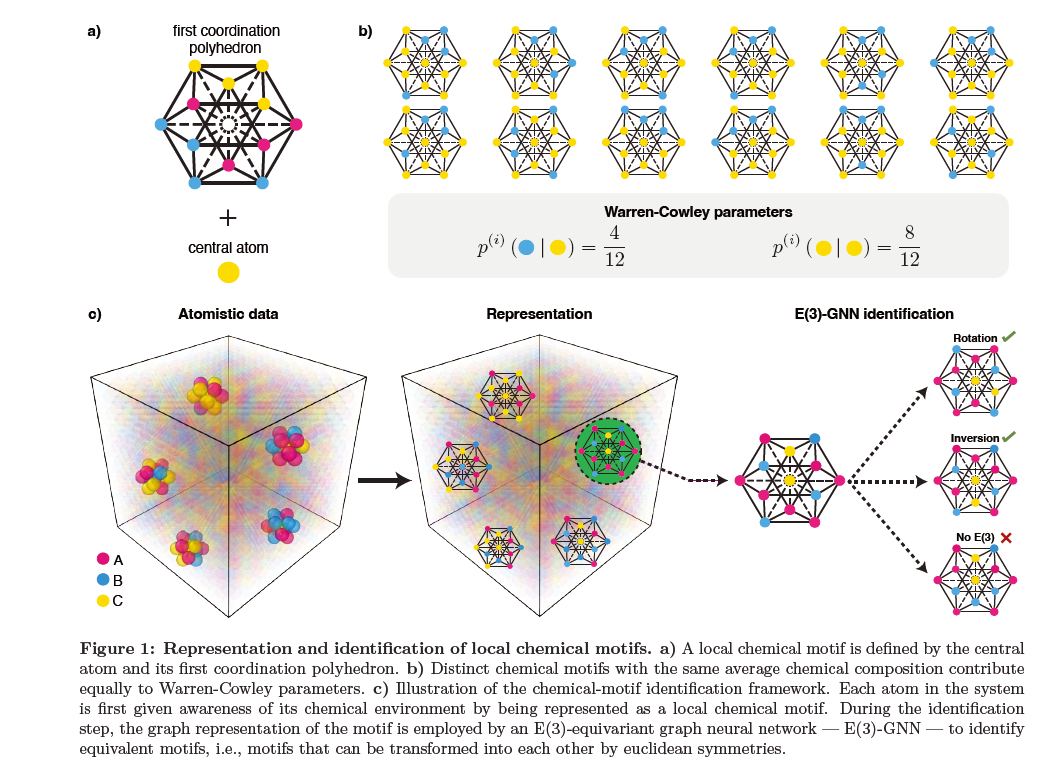
Figure 2
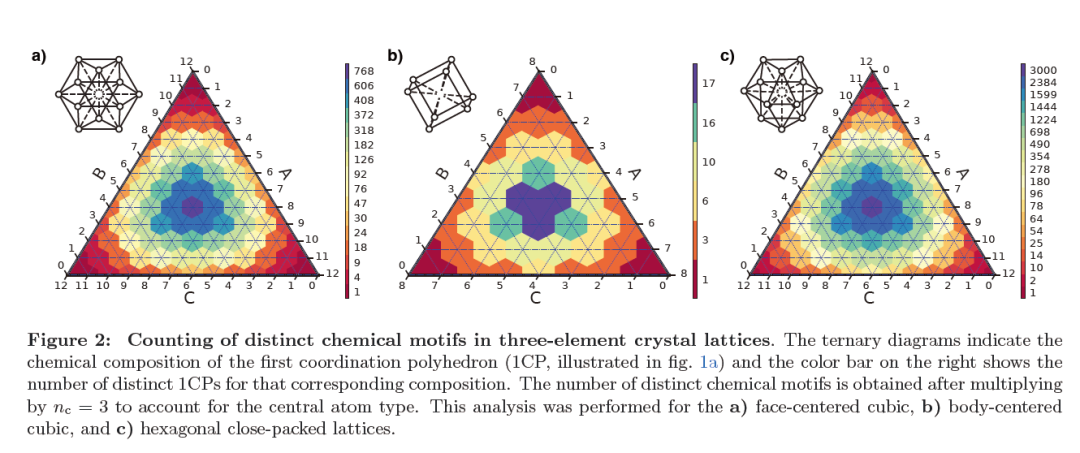
Figure 3
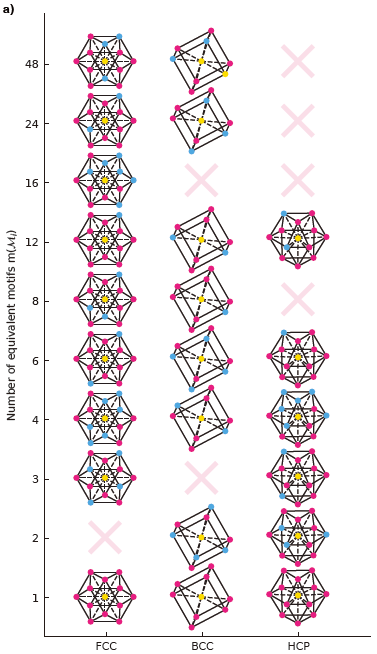
Figure 4
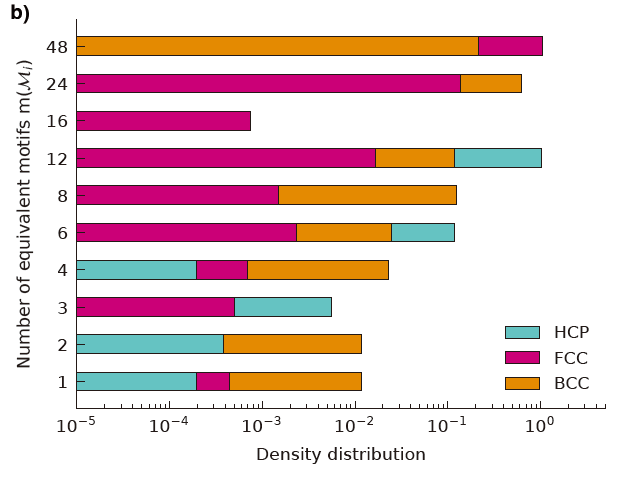
Figure 5
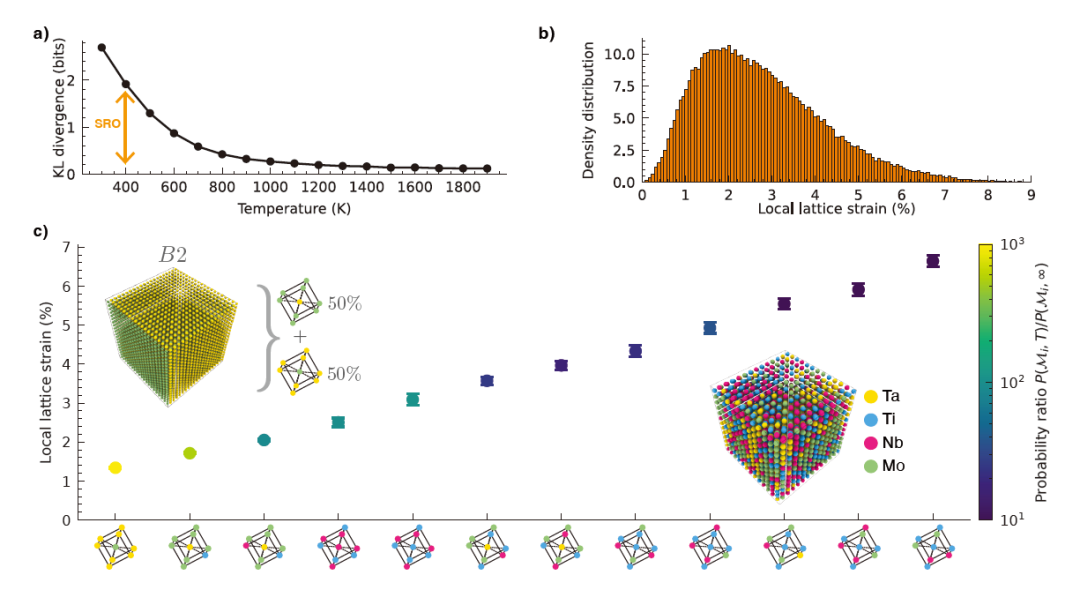
Figure 6
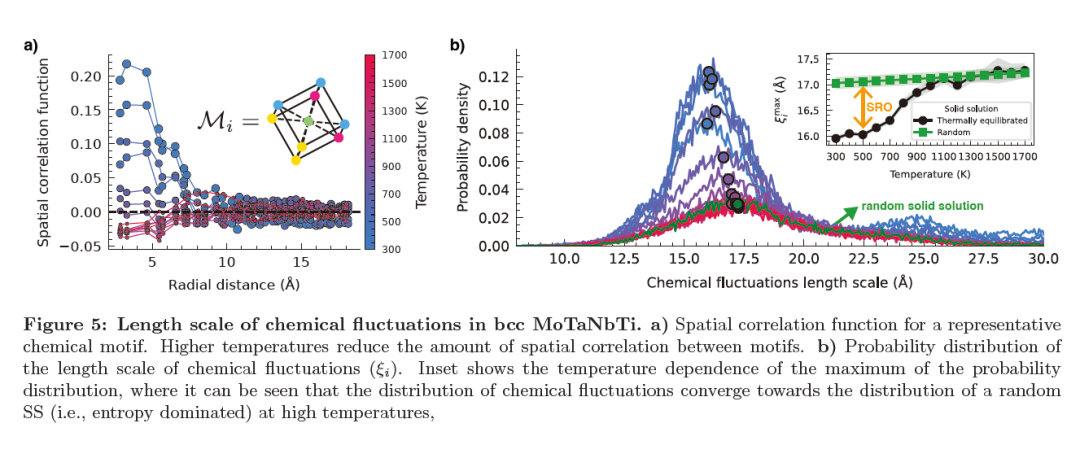
Figure 7
WarrenCowleyParameters
OVITO Python modifier to compute the Warren-Cowley parameters, defined as:
α i j m = 1 − p i j m c j ,
where m denotes the m-th nearest-neighbor shell, p i j m is the average probability of finding a j-type atom around an i-type atom in the m-th shell, and c j is the average concentration of j-type atom in the system. A negative α i j m suggests the tendency of j-type clustering in the m-th shell of an i-type atom, while a positive value means repulsion.
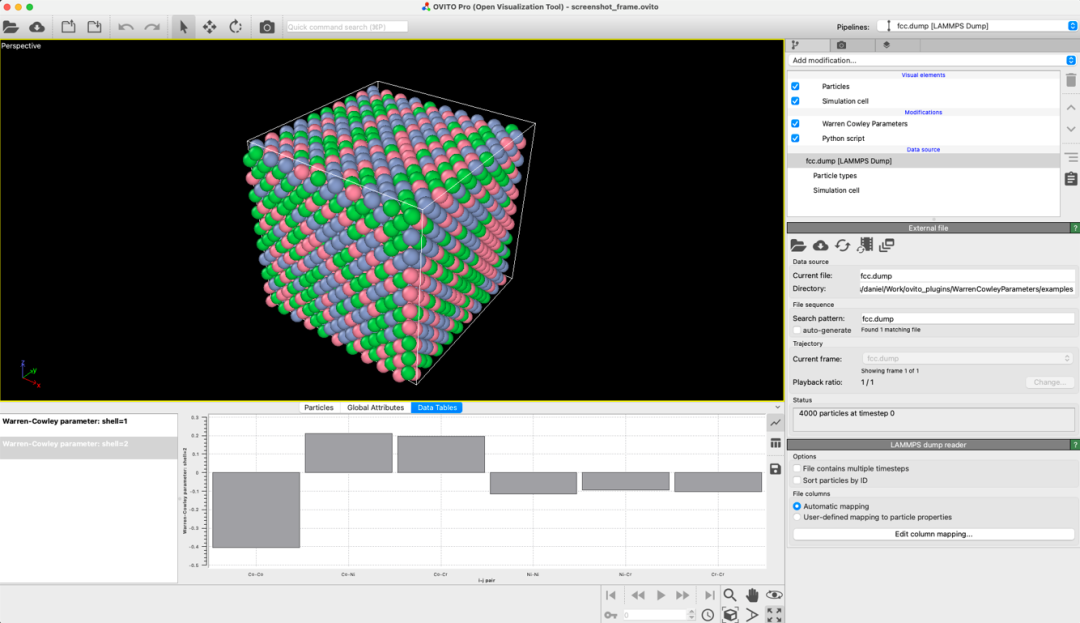
from ovito.io import import_fileimport WarrenCowleyParameters as wcpipeline = import_file("fcc.dump")mod = wc.WarrenCowleyParameters(nneigh=[0, 12, 18], only_selected=False)pipeline.modifiers.append(mod)data = pipeline.compute()wc_for_shells = data.attributes["Warren-Cowley parameters"]print(f"1NN Warren-Cowley parameters: \n {wc_for_shells[0]}")print(f"2NN Warren-Cowley parameters: \n {wc_for_shells[1]}")
Installation
For a standalone Python package or Conda environment, please use:
pip install --user WarrenCowleyParameters
For OVITO PRO built-in Python interpreter, please use:
ovitos -m pip install --user WarrenCowleyParameters
If you want to install the lastest git commit, please replace WarrenCowleyParameters by git+https://github.com/killiansheriff/WarrenCowleyParameters.git.
================================
![]()
以上是我们分享的一些经验或者文章的搬运,或有不足,欢迎大家指出。若留言未回复,重要的消息可以留言再提醒一下,因为超过 48 小时不可回复。
如有侵权,请联系我们立马删除!
👇
文章题目:
Chemical-motif characterization of short-range order with E(3)-equivariant graph neural networks
👇

















 3392
3392

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









