







摘要:在这项研究中,使用MES和scPTZ这两个实验模型测试了3,4-二氢-2(1H)-喹啉酮衍生物在小鼠体内的抗惊厥活性,并测试了神经毒性。此外,还进行了与GABAA受体的体外结合实验。药理实验结果表明,化合物5b表现出最佳的抗惊厥活性,且优于carbamazepine和ethosuximide,并对GABAA受体的结合亲和力最强(IC50 = 0.12μM)。并使用分子对接研究了化合物5b与GABAA受体的结合方式。
分子对接(Molecular docking)是基于结构药物设计的核心模拟手段,依据受体与配体作用时的几何匹配和能量匹配过程,模拟受体-配体相互作用,预测两者间最佳的结合模式和结合亲和力。采用分子对接模拟技术,科研人员可以进行基于结构的药物虚拟筛选,药物分子的结构改造,配体和受体相互作用的机理研究等工作,从而大大提高实验效率。在Discovery Studio这一分子模拟的综合平台中,分子对接程序包含Libdock、CDOCKER、Flexible Docking,这三种对接算法各有优势,能够满足广大科研工作者的多种应用需求,为其提供配体受体间相互识别的“利器”。
抗惊厥类化合物及其与GABAA受体的结合的研究
一 研究背景
癫痫病是人类的主要健康问题,它是由大脑中神经元过度异常放电引起的非自主性抽搐,影响了全世界1%的人口。然而,我们仍无法在细胞水平上确定癫痫的发病机理。目前,药物治疗是癫痫治疗的主要方法。据统计,只有50%的患者可以通过现有的抗惊厥药物完全治愈,大约20%的患者未得到充分治疗并显示与治疗相关的副作用,其余30%是药物无法治愈的顽固性癫痫患者。因此,迫切需要开发高效、低剂量的抗惊厥药。新型靶向抗惊厥药物的开发可能是治疗癫痫的一种非常有效的策略。γ-氨基丁酸A型(GABAA)受体是中枢神经系统(CNS)药物的重要靶标。苯并二氮杂卓类药物位点(BZs-S)是GABAA受体上的重要活性位点,尤其是在抗惊厥药物的开发中起着重要的作用。BZ药物具有共同的化学结构特征:疏水区域A和C,以及距A和C适当距离的受体区域(B)。含吡咯的五元杂环衍生物和3,4-二氢-2(1H)-喹啉酮也被广泛用于抗惊厥药物的研究,并表现出良好的抗惊厥活性。
二 研究过程
在这项研究中,为了找到与BZ相似结构特征的化合物,作者将1,3,4-恶二唑和3,4-二氢-2(1H)-喹啉酮结构片段结合在一起以提高其对癫痫的疗效,设计并合成了一系列化合物5a-j。为了研究化合物中3,4-二氢-2(1H)-喹啉酮片段的重要性,作者以两种方式修饰了该片段,设计合成了6a-j和11a-j一系列衍生物。并使用MES和scPTZ以测试化合物在小鼠体内的抗惊厥活性,并测试了神经毒性。
分子对接是一种主要用于研究分子间相互作用并预测结合方式和亲和力的理论模拟方法,已成为计算机辅助药物研究领域的一项重要技术。为分析先目标化合物与BZs活性位点的可能作用方式,选择了活性最高的化合物5b、地西泮(阳性对照)和GABAA受体的BZs-S (PDB ID:6HUP),使用BIOVIA Discovery Studio2019软件中的分子对接模块用于对接分析。结果显示,BZs-S与化合物5b和地西泮相互作用的主要氨基酸为Tyr58、Phe77、Tyr160、Val203、Ser205、Ser206和Tyr210。化合物5b的对接结果表明,其与BZs-S周围氨基酸的相互作用力高于地西泮。化合物5b和地西泮对接叠加在GABAA受体BZ结合袋中,通过分析化合物5b与地西泮的对接结果,可以推测化合物5b与GABAA受体的相互作用与地西泮与GABAA受体的相互作用相似。另外,并使用DS 2019预测了化合物的理化特性和极性表面积。结果显示目标化合物均能满足Lipinski's五原则、具有较高的口服生物利用度以及良好的ABS和BBB渗透性。总之,通过分子模拟的方法可预测化合物的理化性质及分析配体与受体间相互作用,更好的解释化合物的作用机制。
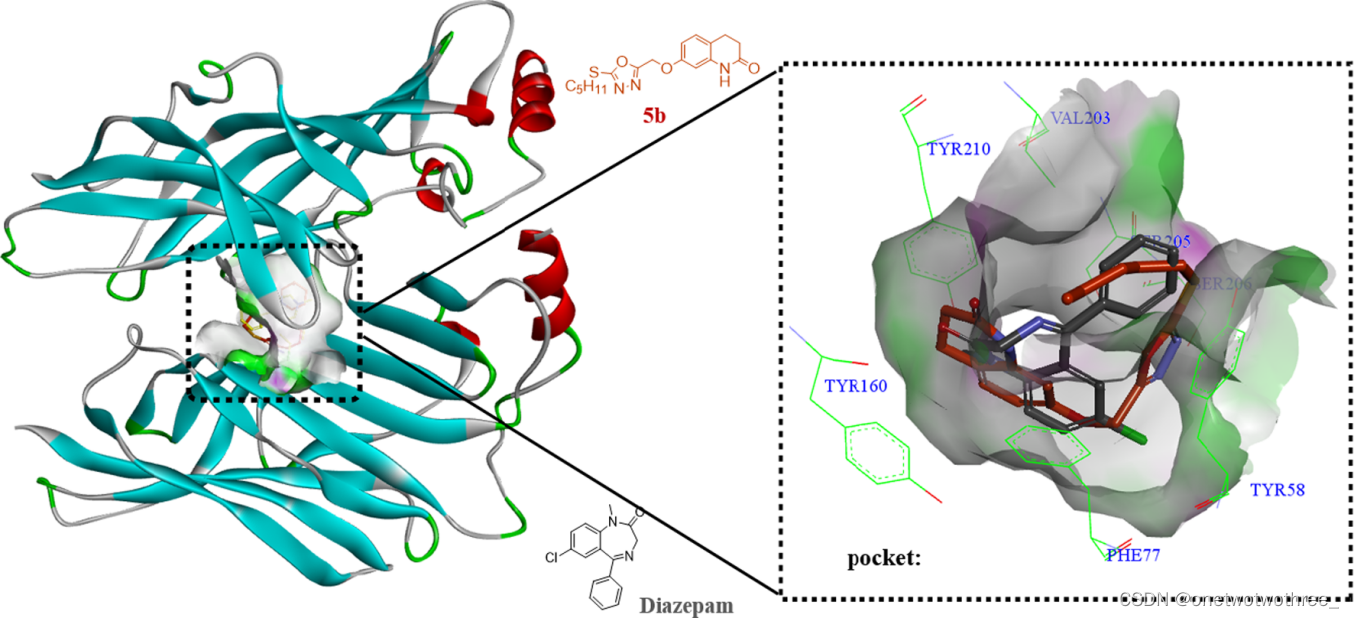
图1 化合物5b(棕色)和地西泮(黑色)叠加在GABAA受体的BZ结合袋中。
MaXFlow分子模拟与人工智能平台
MaXFlow生物医药智能创新平台,由创腾科技自主研发,旨为不同领域的一线创新科技工作者提供一个合作共享的B-S架构平台。以“数据自由,模型自由”为理念,在结构模型与预测模型进行融合的基础上,实现模拟与AI需求的合并,为研发赋能。
- 填补数据产生保存与数据使用赋能断层
- 打通空间结构模型与数据预测模型壁垒
- 合并经典模拟计算与新兴AI预测需求
- 降低背景知识储备与复杂软件使用门槛
通过便捷的网页端操作,可实现大、小分子模型的构建与优化,动力学模拟,分子对接,分子间相互作用展示。小分子药物方面,通过分子性质计算以及多种机器学习与深度学习的方法,在工作流中帮助用户实现数据的挖掘以及相关构效关系的搭建,同时可以通过一键部署的方式实现药代动力学及不同目的的AI预测与共享。对于大分子,基于流行AI模型的运用,更加准确的实现大分子间相互作用预测。多样的APPs为大分子药物研发提供可靠保障。


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









