



本文将为大家分享openEuler用户软件仓(EUR)的使用方法,帮助大家在EUR中构建自己的软件包。
基本概念
在使用用户软件仓前,有如下几个概念需要您了解:
用户:用户是使用本系统的主体,也是所有操作的发起者;
项目:每个用户可以创建多个项目,项目用于组织一个或多个软件包,每个项目可以针对这组软件包生成针对各个openEuler版本的软件包仓库;
软件包:代表一个源码包;
构建:rpm包的一次构建的上下文,包括srpm包和其构建生成的一些rpm包;
仓库:针对特定openEuler版本构建的的软件包仓库。
逻辑如下:
└── User
├── Project
│ └── package 1
│ ├── build 1
│ │ └── log
│ ├── build 2
│ │ └── log
│ ├── build 3
│ │ └── log
│ │ └── rpms
│ │ └── src.rpm
│ └── package 2
│ └── repo for 22.03-x86_64
│ ├── pkg1.noarch.rpm
│ ├── pkg1-debuginfo.rpm
│ ├── pkg2.x86_64.rpm
│ ├── pkg2-debuginfo.rpm
│ └── repo for 22.03-aarch64如何使用
准备工作
使用EUR无需任何门槛,您只需注册一个openEuler账号即可使用。
openEuler 账号中心:
https://id.openeuler.org/zh/profile
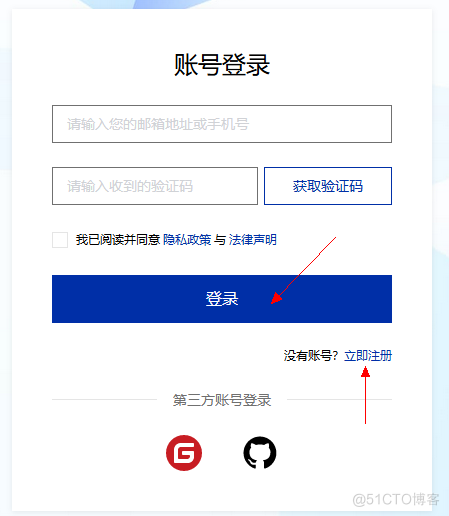
openEuler账号可以通过Gitee或Github账号直接登录,也可以直接通过邮箱直接注册。
进入用户软件仓首页并且登录之后,就可以开始构建自己的软件包了。
用户软件仓首页:
https://eur.openeuler.openatom.cn/
创建一个新的项目
点击new project按钮,创建一个新的项目:
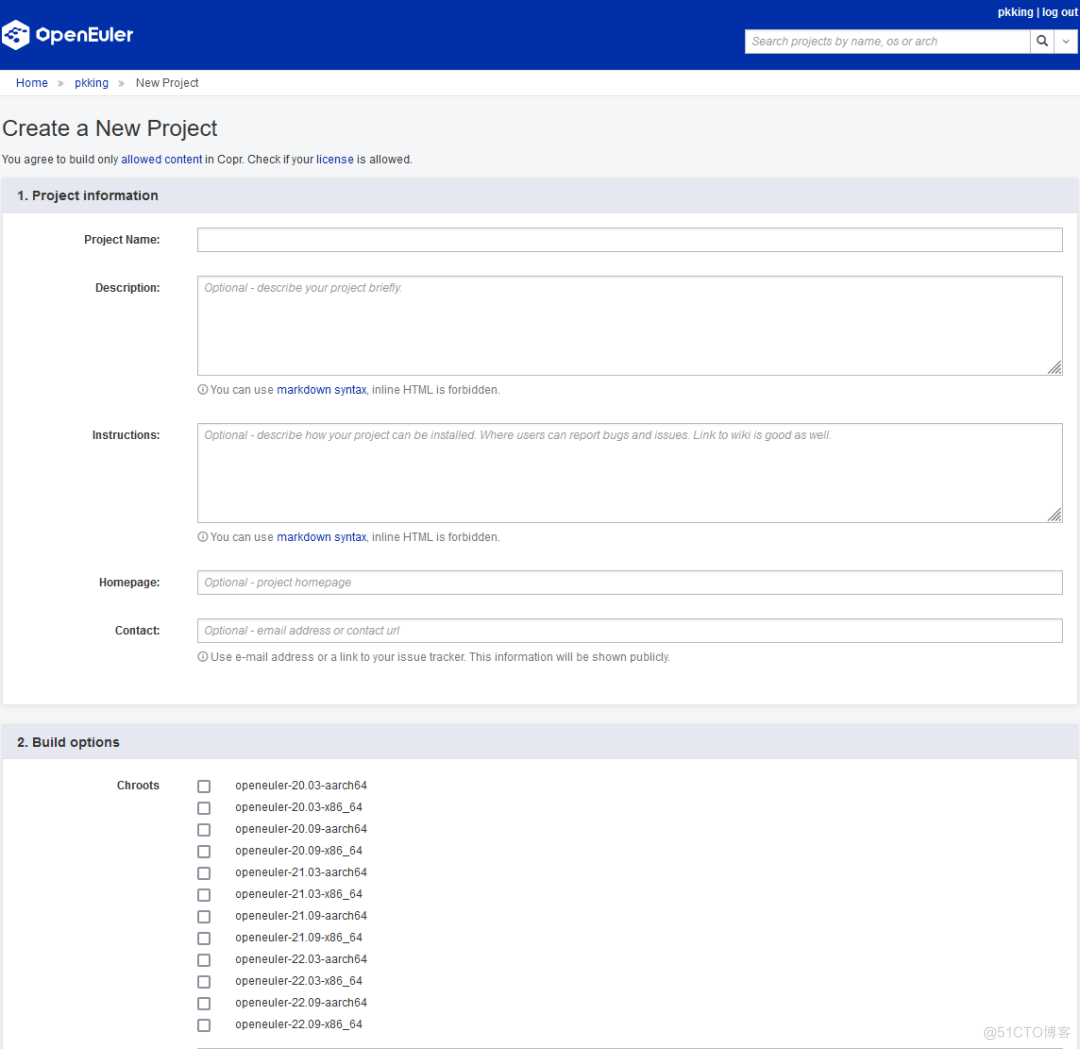
项目名称:后续无法再修改;
项目描述,指南,主页,联系人:可选配置,项目创建后支持修改;
Chroots:必须配置,选择需要的软件包构建环境,项目创建后支持修改;
External Repositories: 可选配置,如果构建过程中依赖其他的软件仓库,可以填写在这里。
项目创建后,在Repo Download处,即可下载对应版本的仓库配置文件。
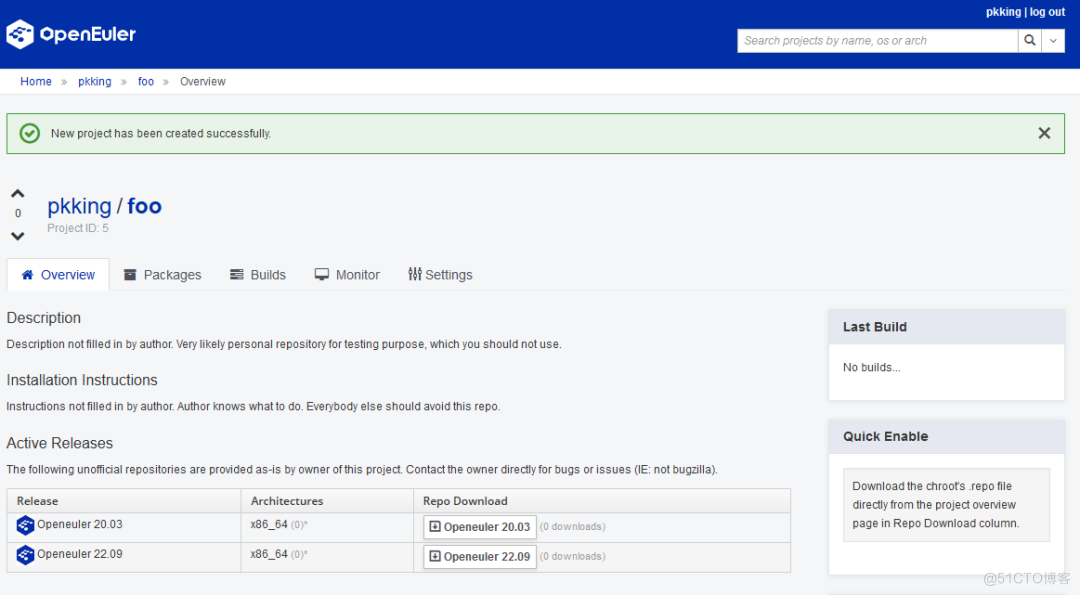
构建一个简单的包
由于当前仓库中可用的软件包较少,开发者们可以自行DIY,添加所需软件包。
点击packages标签页,创建一个新的软件包:
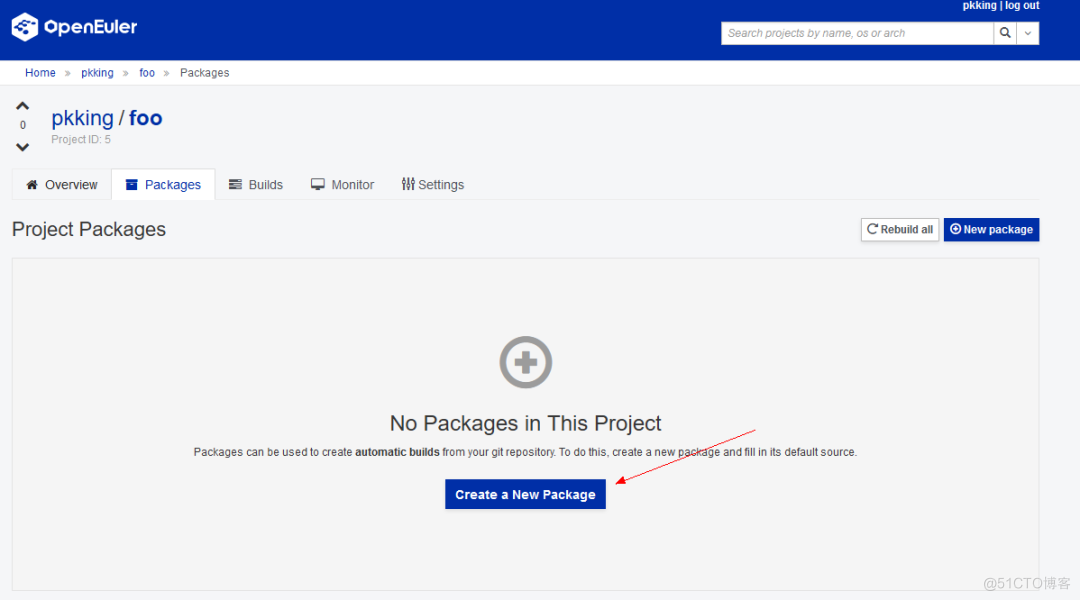
这里我们直接添加openEuler的isulad软件包,因为其已经包含构建所需要的spec和源码包。
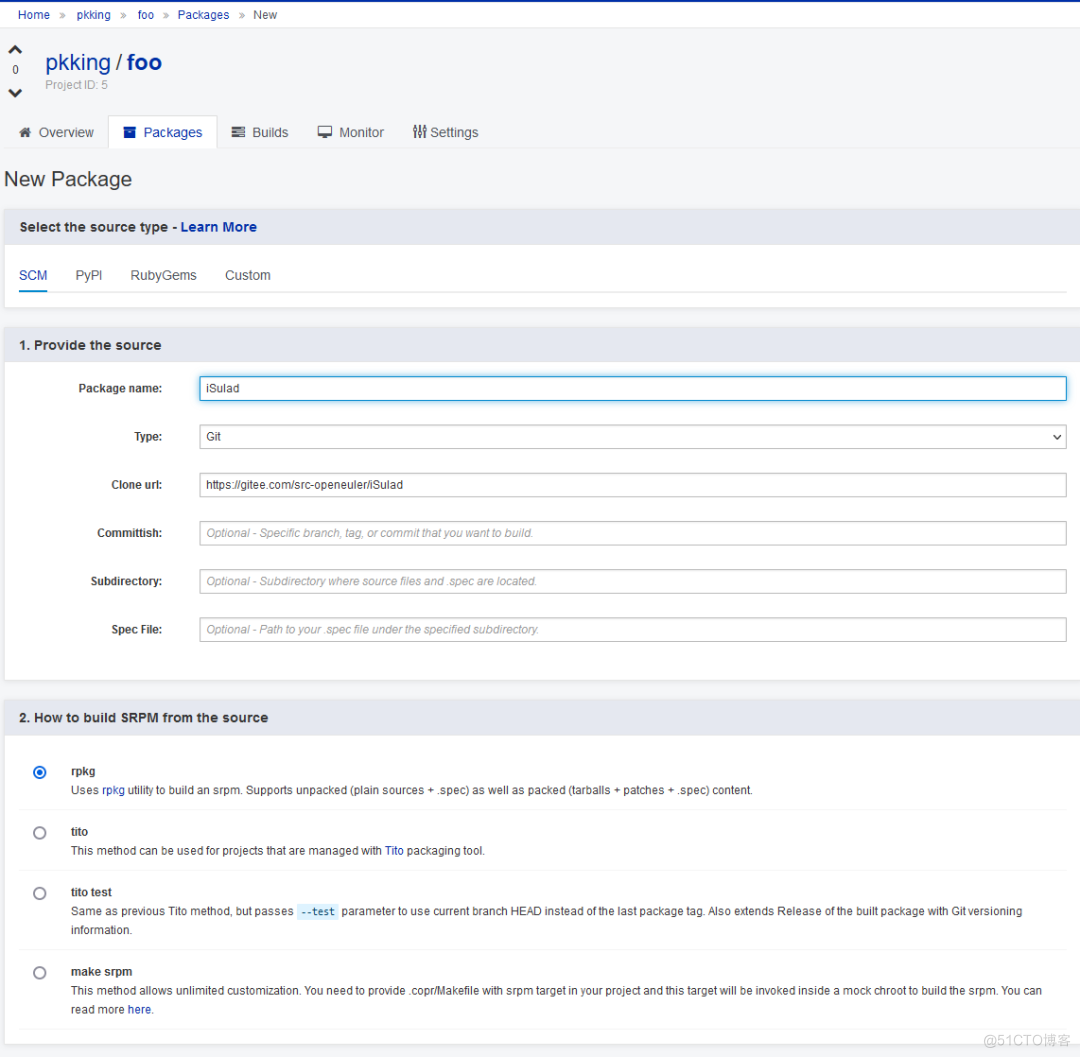
软件包创建完成后,点击rebuild即可触发一次构建。
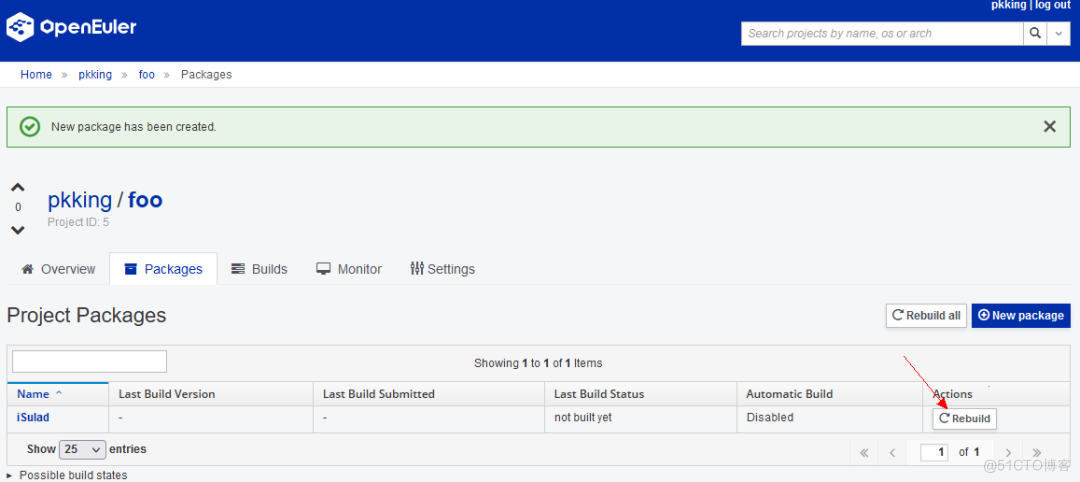
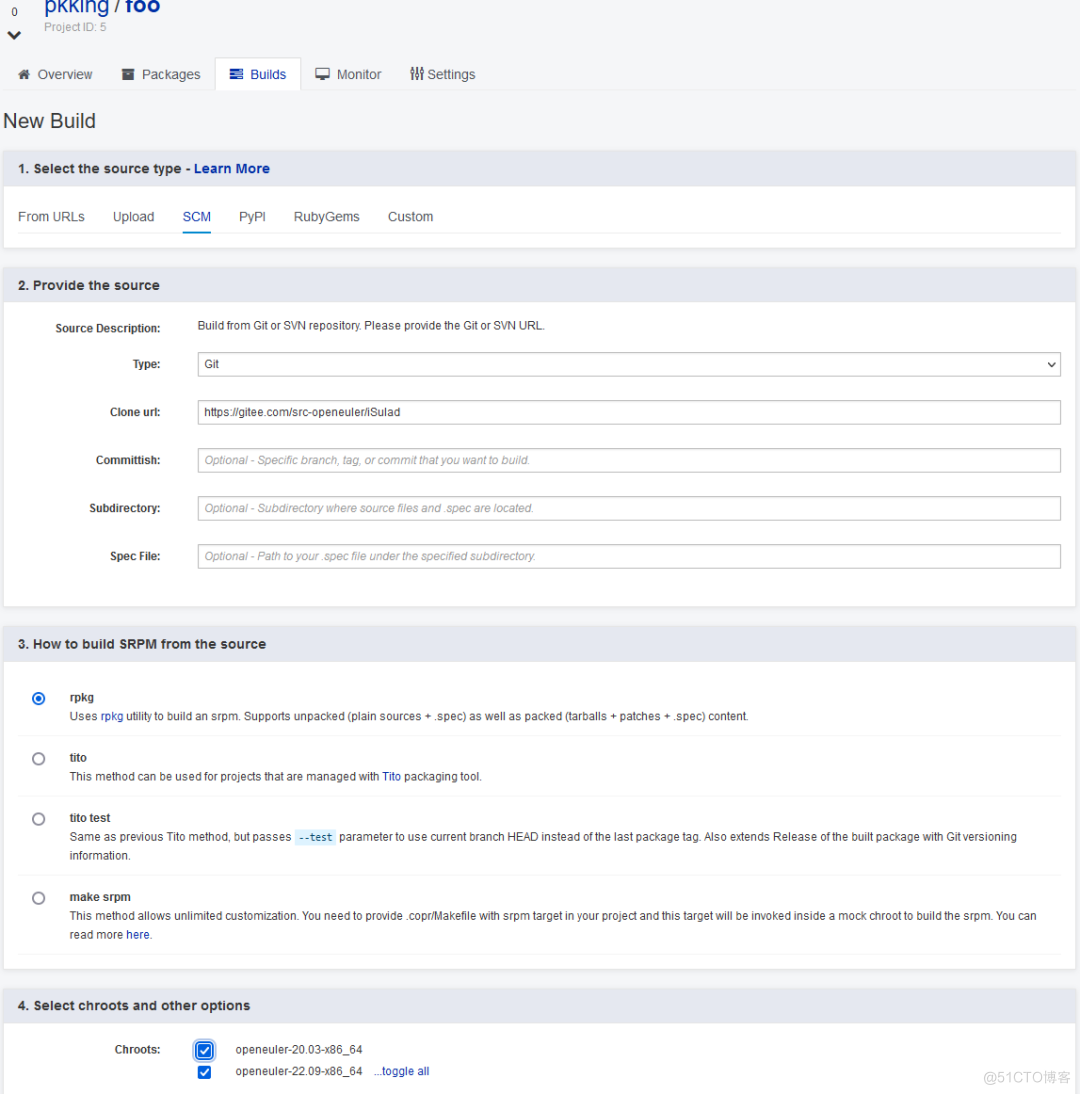
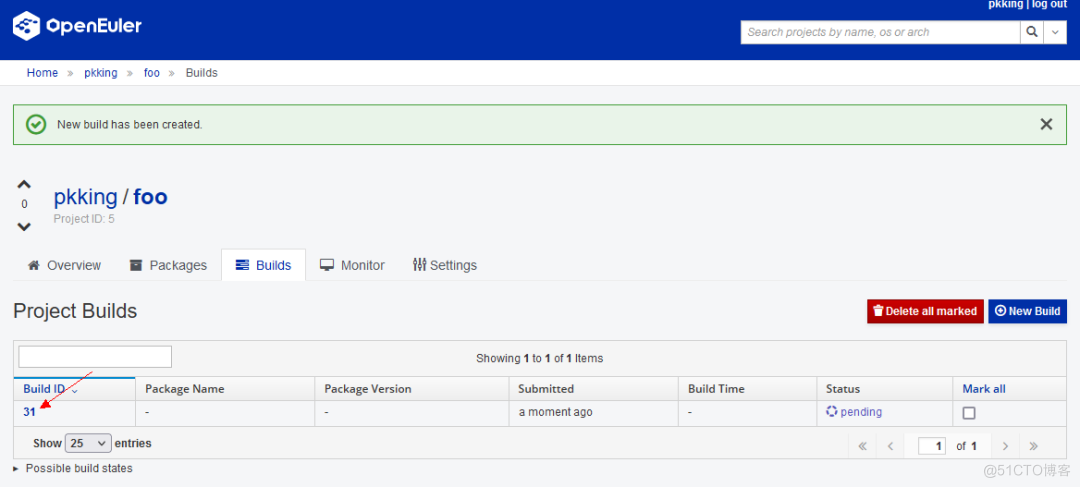
再次点击Build,后台构建系统就会开始构建你的软件包,通过点击任务id,可以实时观看任务的日志。
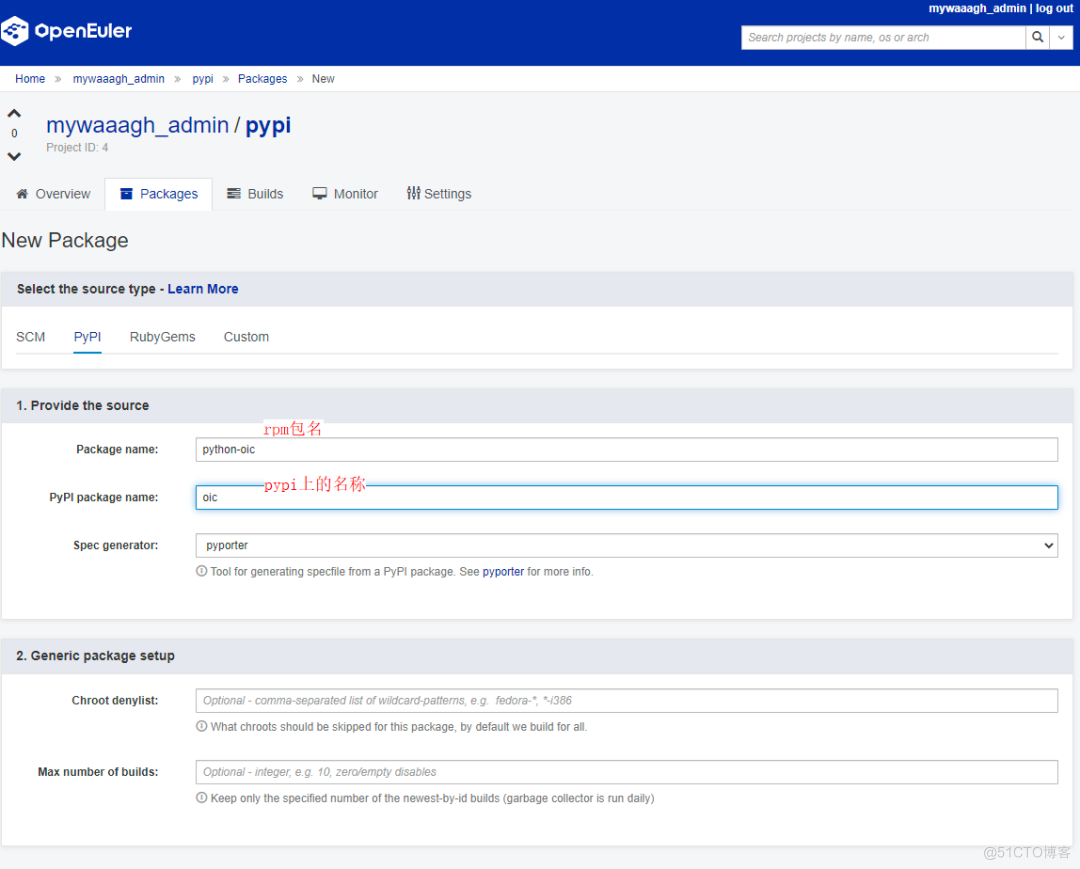
快速构建pypi上的软件包
个人软件仓提供了快速打包pypi上软件包的能力,在添加软件包时,可以直接根据pypi上的包名添加。
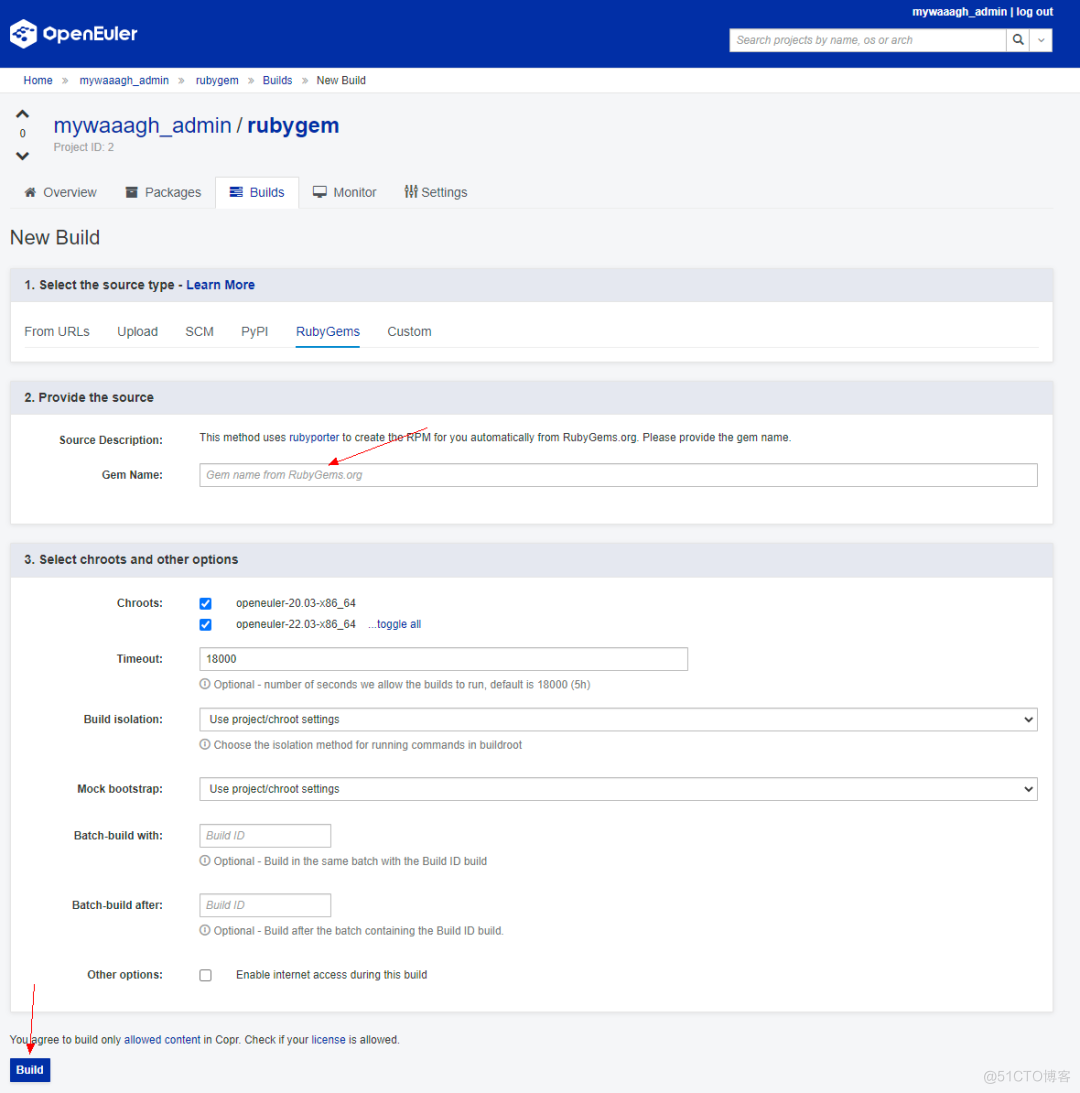
快速构建rubyGem上的软件包
rubyGem上的软件包,可以通过project->builds->new build来进行构建。
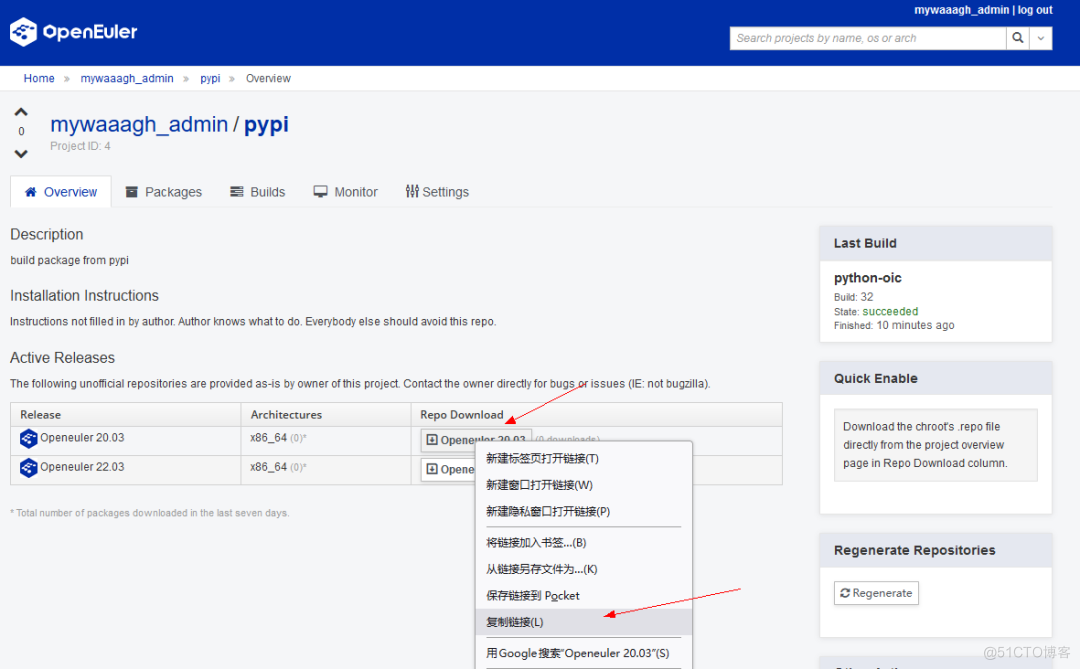
应用EUR中的软件包
curl -OL<下面复制得到的url>,可以直接下载对应的仓库配置;
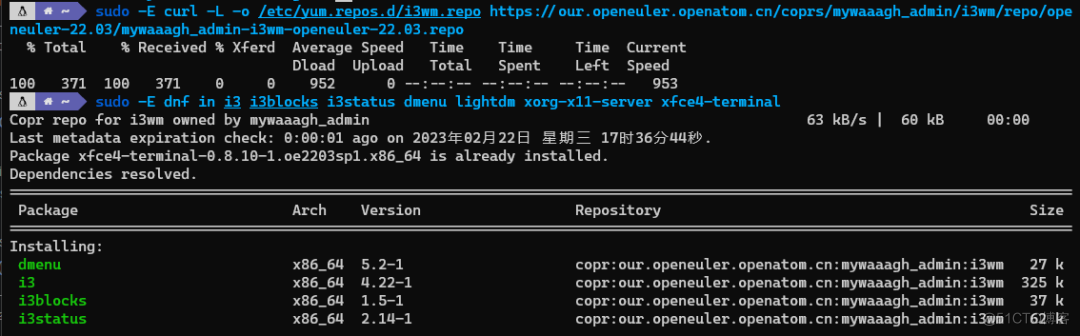
再使用dnf in即可安装对应仓库中的软件包,每个project都有独立的gpg key对rpm包进行签名。


















 1674
1674

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











