









 本文提出scDeepCluster,一种基于单细胞模型的深度嵌入聚类方法,用于单细胞RNA测序(scRNA-seq)数据聚类。该方法结合ZINB模型与聚类损失,能同时学习特征表示和聚类。实验表明,它在模拟和真实数据集上优于现有方法,且在大型数据集上有良好可扩展性。
本文提出scDeepCluster,一种基于单细胞模型的深度嵌入聚类方法,用于单细胞RNA测序(scRNA-seq)数据聚类。该方法结合ZINB模型与聚类损失,能同时学习特征表示和聚类。实验表明,它在模拟和真实数据集上优于现有方法,且在大型数据集上有良好可扩展性。
摘要
单细胞RNA测序(scRNA-seq)有望提供比大量RNA测序更高的细胞差异分辨率。通过scRNA-seq对转录组进行聚类分析,可以揭示细胞的异质性和多样性。然而,scRNA-seq数据的聚类分析仍然是一个统计和计算上的挑战,因为普遍存在的退出事件使数据矩阵与普遍的“假”零计数观测模糊。在这里,我们开发了scDeepCluster,这是一种基于单细胞模型的深度嵌入聚类方法,它通过对scRNA-seq数据生成的显式建模来同时学习特征表示和聚类。基于对来自四个代表性单细胞测序平台的大量模拟数据和真实数据集的测试,scDeepCluster在各种集群性能指标下优于最先进的方法,并表现出更好的可扩展性,运行时间随着样本量的增加而线性增加。scDeepCluster的准确性和效率使其成为一种很有前途的大规模scRNA-seq数据聚类算法。
介绍
单细胞RNA测序(scRNA-seq)可以揭示细胞群的异质性和多样性,并帮助研究人员更好地理解复杂的生物学问题。聚类分析在大多数scRNA-seq研究中都是常规的。聚类是经典的无监督机器学习问题,近几十年来得到了广泛的研究。目前已经提出了许多流行的方法,如k-means、高斯混合模型(gmm)和谱聚类。然而,在scRNA-seq数据集中的细胞群聚类仍然是一个统计和计算上的挑战。与大量RNA-seq数据和微阵列数据相比,聚类scRNA-seq数据的主要问题是它们非常稀疏,以至于大多数测量值都是零(由于低RNA捕获率)。缺失基因测量被定义为遗漏事件,并导致“假”零计数观察。测序平台的最新进展使数千个细胞的通量大幅增加。由于每个细胞的测序深度相对较浅,这些技术特别容易出现dropout事件。此外,scRNA-seq显示出基因表达水平的高度差异,即使在同一组细胞中也是如此。这些技术和生物因素共同引入了大量的变化和噪音,这使得聚类成为一项特别具有挑战性的任务。
最近开发了几种聚类方法来克服这些挑战。例如,Xu和Su提出了SNN-Clip,这是一种利用共享近邻概念的方法。这种策略可以有效地处理高维数据。许多研究使用了涉及迭代聚类的复杂技术。这些方法检测亚型之间的关系,并通过差异基因表达分析进一步验证。DendroSplit是一个可解释的聚类框架,它使用特征选择来揭示scRNA-seq数据中具有生物学意义的多个水平的种群。Wang和他的同事利用多核学习(SIMLR)进行单细胞解释。本质上,SIMLR是一种谱聚类方法,它结合多个核来学习最适合数据结构的鲁棒距离度量。为了表征scRNA-seq数据的稀疏性,Park及其同事还提出了一种多核谱聚类方法,但建议通过L1惩罚来施加稀疏结构(MPSSC)。这两种最新的方法代表了当前最先进的scRNA-seq数据聚类方法。尽管这些基于光谱聚类的方法表现出了良好的性能,但它们存在两个问题。首先,它们依赖于完整的图拉普拉斯矩阵,这在计算和存储方面非常昂贵。拉普拉斯矩阵的计算和存储通常在数据点(这里的单元格数)的数量方面具有二次或超二次复杂性,并且矩阵的分解需要三次复杂性。这些复杂性表明谱聚类存在严重的可伸缩性问题。在许多scRNA-seq研究中,通常需要聚集数千个细胞,这需要具有巨大内存的超级计算机。例如,参考文献16中使用一台具有800Gb内存的机器来对数千个单元进行集群。这限制了基于光谱的聚类方法在大型scRNA-seq数据集(现代scRNA-seq平台的典型输出)上的应用,因为它们通常同时分析数千甚至数万个细胞。其次,光谱聚类方法没有明确地模拟scRNA-seq计数数据的过分散和零膨胀等特征,留下了进一步改进的空间。
另一项相关的研究侧重于计算scRNA-seq数据中由于缺失而导致的缺失值(假零)。这样的推算有望改善各种下游分析,包括聚类。最近在这方面开发了几种方法和工具,包括统计模型CIDR(通过输入和降维聚类),scImpute, MAGIC和SAVER,仅举几例,以及深度学习方法DeepImpute, DCA(深度计数自动编码器)和scScope。CIDR是一种快速的类似pca的算法,它考虑了辍学。它采用一种简单的隐式归算方法来减轻scRNA-seq数据中dropouts的影响,然后基于前几个主坐标进行聚类。DeepImpute应用标准的深度神经网络,使用具有足够读覆盖率的高度相关基因作为输入来预测目标基因的缺失值。scScope和DCA都是基于自动编码器的。自编码器是一种以无监督方式学习有效特征表示的深度神经网络(dnn)。
scScope实际上重复运行常规自动编码器三次,使用前一次运行的最终输出作为下一次运行的初始输入。与常规自编码器相比,DCA提出用基于零膨胀负二项(ZINB)模型的损失函数代替传统的均方误差(MSE)损失函数,以更好地表征scRNA-seq数据。我们已经使用ZINB模型对微生物组测序数据进行差异表达分析26,并表明它可以有效地表征离散、过度分散和零膨胀的计数数据。DCA的结果还表明,使用ZINB对DCA进行估算可以改善多种典型下游scRNA-seq数据分析。这些改进表明,ZINB模型在描述普遍的辍学事件(scRNA-seq数据中的主要统计挑战)方面是有效的。
然而,这些专注于输入的方法并没有针对聚类进行设计和优化,尽管我们可以作为一种朴素的解决方案,首先输入scRNA-seq数据,然后使用k-means等简单聚类。这种划分策略对于聚类来说不是最优的,正如我们的方法与DCA的比较所示(scScope不共享它们的程序)。
由于“维度诅咒”,聚类在小维度上的表现要比在高维度上好得多。深度神经网络是将原始高维数据映射到小潜在空间的非线性转换函数参数化的自然选择。深度神经网络已经证明了理论上的函数逼近能力和特征学习特性。最近的研究表明,深度学习可以成功地实现。当应用于图像和文本数据集时,在聚类任务上具有良好的性能。同时,一项研究表明,dnn可以以监督的方式降低scRNA-seq数据的维数。相比之下,最近发表的一种深度生成模型scvis以无监督的方式捕获和可视化scRNA-seq数据中的低维结构。
因此,我们提出了一种深度学习聚类方法,该方法将ZINB模型与聚类损失有原则地结合在一起(其架构见图1)。我们的方法旨在显式优化聚类,同时执行降维。在提出的框架中,将scRNA-seq数据的读取计数矩阵映射到低维潜在表示的非线性函数由基于ZINB模型的自编码器学习,而潜在空间上的聚类任务则由“深嵌入聚类”(DEC)算法中描述的Kullback-Leibler (KL)散度聚类完成。我们还将去噪自编码器技术引入到基于ZINB模型的自编码器中,使其能够更好地学习数据的鲁棒特征表示。我们将这种方法命名为“基于单细胞模型的深度嵌入聚类”。使用模拟和真实的scRNA-seq数据,我们证明scDeepCluster比竞争的最先进的聚类方法带来了显着的准确性提高。此外,我们表明scDeepCluster比竞争方法需要更少的内存和时间复杂度。这种吸引人的计算效率使得scDeepCluster更适合于分析大型scRNA-seq数据。
仿真评估 Simulation evaluation of scDeepCluster
为了评估scDeepCluster在scRNA-seq聚类分析中的性能,我们在接近不同生物场景的广泛设置下设计了以下模拟。具体来说,我们使用R软件包Splatter来模拟scRNA-seq读取计数数据。我们模拟了1500个细胞,每个细胞有2500个基因,形成大小相同的三组(每组500个细胞)。我们使用以下三个性能指标来评估获得的聚类与真实标签之间的一致性:归一化互信息(NMI)、聚类精度(CA)和
调整后兰特指数(ARI)。NMI和CA的取值范围为0 ~ 1,而ARI可以产生负值。这三个指标是对两个聚类标签一致性的统计;数值越高,聚类的一致性越高。我们在相同的环境下重复所有实验20次。将scDeepCluster与DCA+k-means、两种多核谱聚类方法MPSSC和SIMLR、CIDR、PCA+k-means、scvis+k-means和DEC进行了比较。
我们首先研究了这八种方法在不同辍学率下的性能,辍学率的定义是表达基因在读取计数中被敲除的比例。为此,我们改变dropout logistic函数的中点参数,生成不同辍学率(12±0.3%,17±0.4%,23±0.5%和30±0.6%)的数据集。NMI的聚类精度(每种设置中20次重复的平均值)如图2a所示(CA和ARI的数据在补充图1中提供)。我们注意到几个有趣的发现。首先,对于所有的方法,准确率随着辍学率的增加而下降,这证实了我们的猜测,即辍学事件使聚类具有挑战性。特别是,PCA+k-means和DEC的性能随着辍学率的增加而急剧下降,但DCA的下降并不明显。DCA的改进证实了对scRNA-seq数据特征的特殊建模可以增强标准DEC模型。其次,scDeepCluster在所有辍学率设置下均显著优于其他方法(p<0.01,单侧配对t检验)。第三,scDeepCluster对不断增加的辍学率具有更强的鲁棒性。scDeepCluster的聚类性能在辍学率增加到30%之前是完美的(NMI≈1),但在此之后仍然保持良好(NMI≈0.9)。相比之下,其他方法的性能随着辍学率的增加而开始迅速而显著地恶化。这些优越的性能表明scDeepCluster可以有效地表征scRNA-seq数据中的辍学事件。实际上,ZINB损失函数不仅有助于scDeepCluster提高聚类性能,而且使其性能更加稳定(Supplementary Fig. 2)。
接下来,我们评估了不同聚类信号强度下的性能。为了做到这一点,我们改变了细胞类型和组之间基因表达的倍数变化水平,这可以通过调整Splatter使用的对数正态分布的方差参数sigma来控制,以产生不同的差异表达强度。对数正态分布的方差参数越大,说明不同聚类样本之间的距离越大,聚类信号越强。因此,将dropout率固定为17% (Splatter的默认设置),我们在不同的sigma值下评估方法。其性能如图2b和补充图3所示。这些模拟数据集的t-SNE39图证实,聚类信号强度随着sigma值的增加而增加(Supplementary Fig. 4)。我们可以再次看到,scDeepCluster在所有不同的聚类信号强度下都显著优于其他方法(p<0.01,单侧配对t检验)。不足为奇的是,不同方法的聚类性能通常随着信号的增加而提高。值得注意的是,当信号增加到弱信号(sigma=0.2)以上时,scDeepCluster的性能显著提高,当sigma增加到0.225时,迅速达到良好的聚类(NMI≈0.86)。随着西格玛的增加,MPSSC的改善是稳定的,但非常小,表明它缺乏利用增加的信号的效率。所有信号的SIMLR失败(NMI接近0)。随着sigma的增加,DCA、PCA、scvis和DEC的聚类效果也有所改善,但DCA和scvis的改善速度要快于PCA和DEC。在这些模拟数据集上,我们发现scDeepCluster对嵌入空间进行k-means初始化后的聚类效果更好,突出了深度学习聚类阶段的贡献(补充图5)。
最后,我们研究了scDeepCluster在三组不平衡样本量下的性能。在参考30之后,我们在将1,500个细胞分配给三组时,通过改变最小保留率rmin(补充说明),产生了不同的不平衡水平。简而言之,rmin越小,不平衡就越严重,最大群体的预期样本量是最小群体的1/rmin倍。将退出率固定为17%,并设置sigma=0.4 (Splatter推荐的默认值),我们将最低留存率从0.1调整为0.9。如补充图6所示,scDeepCluster的性能相当稳健。在rmin减小到0.3时,它仍保持完美的聚类一致性(NMI≈1),即使在极不平衡的情况下(rmin=0.1),其性能仍保持良好(NMI>0.85)。
Application to real data
我们将scDeepCluster应用于四个真实的scRNA-seq数据集来验证其性能。这四个数据集来自四个代表性的测序平台:来自10X基因组学平台的PBMC 4k细胞(10X PBMC),来自液滴条形码平台的小鼠胚胎干细胞(小鼠ES细胞),来自Microwell-seq平台的小鼠膀胱细胞(小鼠膀胱细胞)。
和来自sci-RNA-seq平台的蠕虫神经元细胞(蠕虫神经元细胞),如表1所示。4个数据集每个样本的细胞数分别为4271、2717、2746和4186个,预处理后的基因数分别为16,449、24,046、19,079和11,955个,每簇形成8、4、16和10组。方法的“真实数据”部分提供了数据集的详细描述。基于谱的聚类方法(MPSSC, SIMLR)具有二次空间复杂度,我们无法在更大的内存(例如256G)下运行它们。为了进行比较,我们从每个数据集中随机抽取了2100个单元格。值得注意的是,随机抽样没有丢弃任何数据集中的任何组。聚类性能的三个指标(NMI、准确率和ARI)如图3a所示。我们观察到,所提出的深度学习聚类scDeepCluster在所有四个数据集上都优于所有其他方法,包括MPSSC, SIMLR, CIDR和scvis。我们使用t-SNE图可视化了四个数据集嵌入呈现的进展(补充图7)。在四个真实数据集上也观察到k-means初始化后性能的提高。
所提出的scDeepCluster模型中的潜在空间是高维输入数据的理想低维嵌入表示。为了说明潜在空间的表示有效性,我们使用t-SNE将scDeepCluster模型中基于ZINB模型的自编码器学习到的最终嵌入点可视化到二维(2D)空间中。还绘制了其他方法的二维空间表示。图3b显示scDeepCluster很好地分离了四种不同白血病抑制因子(LIF)戒断间隔(0,2,4和7天,以不同颜色突出显示)的小鼠ES细胞,只有少数红色样本与蓝色混合,少数绿色样本与黄色混合。相比之下,绿色和黄色样品与其他竞争方法混合在一起。对其他三个数据集进行了类似的观察,其中相同类型的细胞在scDeepCluster的低维嵌入表示中被很好地分离,只有一些异常值,并且比竞争方法要好得多。看到这些方法在非常大的scRNA-seq数据集(例如数万个细胞)上的性能是很有趣的。为此,我们因此增加了两个额外的真实数据集,分别有27,000和68,000个单元格(补充表1)。SIMLR和MPSSC未能运行这两个大数据集。我们观察到scDeepCluster与其他竞争方法相比具有类似的优势(补充图8)。
scDeep集群的可扩展性
从业者现在正面临着越来越多的细胞在scRNA-seq实验。因此,建立一种能够处理大型数据集的分析方法变得至关重要。为了评估scDeepCluster的可扩展性,我们总结了scDeepCluster在不同样本量数据集上的运行时间。我们模拟了一个由10万个细胞组成的大数据集,其中有3000个基因,这些细胞中有10个组,并将这个数据集从5000个降至10万个细胞(5,000,7,500,10,000,25,000,50,000,75,000,100,000)。图4a报告了scDeepCluster在这些下采样数据集上的预训练和聚类阶段的运行时间。我们可以看到,与谱聚类的二次运行时间复杂度不同,scDeepCluster的运行时间随细胞数线性扩展。这样的计算效率使scDeepCluster成为分析大型scRNA-seq数据集的一个非常有吸引力的工具。此外,我们发现scDeepCluster的聚类性能对不同样本量具有相当强的鲁棒性(图4b)。
Discussion and conclusion
在大多数聚类研究中,我们假设k(聚类的数量)是在竞争算法之间的比较中给出的。在实践中,这些信息通常是未知的。当所使用的k与实际集群数量之间存在很大的不匹配时,可能会导致模型不匹配问题。需要一种确定最优簇数的方法。为此,在其他聚类方法30之后,我们可以通过引入度量泛化性来采用肘形方法策略(G,补充说明)。简单地说,我们将数据分成训练集和验证集,并设置G为训练和验证聚类损失之间的比率。我们计算了不同簇数k下的G。我们观察到,当k大于最优簇数时,G急剧下降,这表明G可以作为估计最优簇数的良好指标(Supplementary Fig. 12)。此外,在聚类阶段,我们在自编码器中使用重构损失函数作为数据相关的正则化项。这样的正则化项可以帮助防止深度嵌入函数的过拟合。一般来说,聚类方法可以分为两大类:判别聚类算法(如谱聚类)和生成聚类算法(如我们的方法)。人们一直认为,训练歧视性模型可能会受到影响
过度拟合。我们的实证实验也表明,光谱聚类模型SIMLR错误地估计了模拟数据集中的聚类数量(Supplementary Fig. 13)。
总之,我们提出了scdeepcluster——一种基于模型的深度学习方法,用于scRNA-seq数据的聚类分析。scDeepCluster可以学习一种潜在的嵌入式表示,该表示以非线性方式优化了高维输入的聚类。特别是,我们明确地使用参数模型来模拟scRNA-seq数据生成,该模型适用于描述带有过多零的计数数据。仿真研究和实际数据应用都表明,将深度学习与这种参数化相结合可以有效地捕获普遍的辍学事件,这是scRNA-seq数据分析面临的主要挑战。相比之下,先前最先进的谱聚类方法(MPSSC和SIMLR)依赖于多个高斯核,这在描述稀疏计数数据时被证明效果较差。通过与几种竞争方法的比较,我们证明了scDeepCluster在模拟和真实数据集上的优越聚类性能。我们还演示了scDeepCluster在大型数据集上的出色可扩展性。随着越来越多的大规模scRNA-seq数据集变得可用,我们期望我们的方法有更多的应用。
Methods
读取计数数据预处理。
原始的scRAN-seq读取计数数据由Python包SCANPY进行预处理。首先,任何细胞中没有计数的基因被释放。其次,计算大小因子,并将读取计数按库大小进行标准化,因此单元格之间的总计数是相同的。在形式上,如果我们表示cell i的library size(总读取计数的数量)为si,那么单元格i的大小因子为si/中位数(s)。最后一步是对读取计数进行对数变换和缩放,使计数数值遵循单位方差和零均值。预处理的读计数矩阵被作为我们基于去噪ZINB模型的自动编码器的输入。
去噪基于ZINB模型的自动编码器。
自动编码器是一种人工神经网络,用于以无监督的方式学习有效的特征表示25。去噪自动编码器是一个自动编码器,它接收损坏的数据点作为输入,并被训练来预测原始的未损坏的数据点作为其输出34。自动编码器通常有一个低维的瓶颈层来学习一个潜在的特征表示。去噪自动编码器被证明在学习数据的鲁棒表示方面更强大,因为它能够学习被输入中小的不相关变化所破坏的输入的表示。在这里,我们应用去噪自动编码器技术,将读取计数的输入映射到一个嵌入的空间来进行聚类。在实际应用中,我们首先用随机高斯噪声破坏输入,然后构造具有规则全连通层的自动编码器。形式上,输入X被噪声破坏
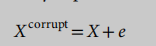 式中,e表示随机高斯噪声。请注意,噪声可以被合并到编码器的每一层中,它被定义为堆叠去噪自动编码器35。我们将编码器函数定义为
z
=
f
W
(
X
c
o
r
r
u
p
t
)
z=f_W(X^{corrupt})
z=fW(Xcorrupt) 和解码器函数
X
′
=
g
W
′
(
z
)
X'=g_{W'}(z)
X′=gW′(z)。编码器和解码器函数都是完全连接的神经网络与整流器激活。这里的W和W‘是函数的学习权值。去噪自编码器的学习过程使损失函数最小化
式中,e表示随机高斯噪声。请注意,噪声可以被合并到编码器的每一层中,它被定义为堆叠去噪自动编码器35。我们将编码器函数定义为
z
=
f
W
(
X
c
o
r
r
u
p
t
)
z=f_W(X^{corrupt})
z=fW(Xcorrupt) 和解码器函数
X
′
=
g
W
′
(
z
)
X'=g_{W'}(z)
X′=gW′(z)。编码器和解码器函数都是完全连接的神经网络与整流器激活。这里的W和W‘是函数的学习权值。去噪自编码器的学习过程使损失函数最小化
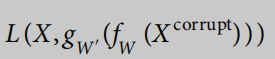
为了捕获scRNA-seq数据的特征,我们使用了一个基于ZINB模型的自动编码器23,而不是一个常规的自动编码器,它被训练来试图重建其输入。与常规的自动编码器不同,基于ZINB模型的自动编码器的损失函数是一个ZINB分布的可能性。ZINB用于表征scRNA-seq中的辍学事件。形式上,ZINB参数化为均值(μ),负二项分布的离散度(θ)和一个附加系数(π),它表示零概率点质量的权重(辍学事件的概率):
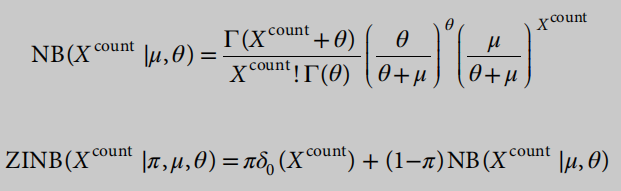 我们在D上添加三个独立的全连接层来估计参数
我们在D上添加三个独立的全连接层来估计参数
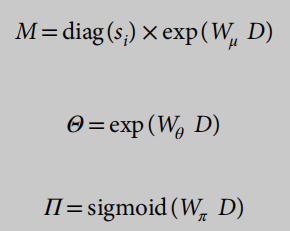
辍学概率在0-1的区间内,因此s型是激活函数的合适选择。基于ZINB模型的自动编码器的损失函数是ZINB似然的负对数:
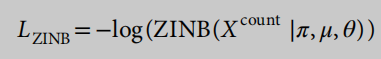
具有局部结构保存的深度嵌入式聚类。
聚类阶段遵循深度嵌入式聚类。考虑将一组n个细胞 X 与每个样本 xi(xi表示d个基因的读计数)聚为k个聚类的问题。深度嵌入聚类不是直接聚类到数据空间X中,而是首先找到一个非线性映射
f
w
x
i
→
z
i
f_w~x_i→z_i
fw xi→zi,其中
Z
Z
Z 为潜在特征空间,
Z
Z
Z 通常比
X
X
X 小得多。聚类将应用于潜在空间
Z
Z
Z。
为了获取单细胞RNA-seq数据的特征,我们应用基于去噪的ZINB模型的自编码器来学习X到z的非线性映射,聚类算法与参考文献中相同。30,定义为分布P和Q之间的Kullback-leibler(KL)散度,其中Q是由学生t分布测量的软标签分布,P是由Q推导出的目标分布。形式上,聚类损失为

我们在聚类阶段之前对基于堆叠去噪的基于ZINB模型的自动编码器进行了预训练。在对基于去噪ZINB模型的自编码器进行预训练后,在嵌入式特征空间中通过标准k-means聚类得到聚类中心的初始化。
因此,该模型有两个组成部分:基于去噪的ZINB模型的自动编码器和聚类部分。然后,该中心的深度集群的目标函数为
Optimization.
通过随机梯度下降(SGD)和反向传播共同优化了DNN和聚类中心{μj }。聚类损失Lc相对于嵌入点zi和聚类中心μj的梯度计算为

实现。
scDeepCluster是在Python 3中使用带有TensorFlow后端的Keras实现的。随机高斯噪声由Keras层“GaussianNoise”实现,噪声电平(参数“stddev”)设置为2.5(噪声的影响见补充图9)。基于ZINB模型的去噪自编码器首先通过Adam的优化器AMSGrad变体进行400次预训练(对于模拟数据,我们预训练了600次),初始学习率lr=0.001, β1=0.9, β2=0.999。聚类阶段的优化器为Adadelta, lr=1.0, rho=0.95。编码器中隐藏全连接层的大小设置为(256,64),解码器与编码器相反,瓶颈层(潜在空间)的大小为32。系数γ的选择为1 (γ的选择见补充图10)。预训练和聚类的批大小为256。聚类的收敛阈值是每批聚类标签增量的0.1%。所有实验均在Nvidia Tesla P100 (16G)上进行。
竞争的方法。
DCA23、SIMLR15、MPSSC16、CIDR18、PCA+k-means、scvis33和DEC30作为竞争方法。DCA通过使用作者的API函数(https://github.com/theislab/dca)直接执行。DCA不是为集群而设计的。因此,我们首先应用DCA(使用作者给出的默认参数)对原始读计数数据进行降噪(计算丢弃计数),然后通过主成分分析(PCA)将高维降噪的读计数矩阵降至二维空间。对投影的二维空间进行k-means聚类。这种方法被称为“DCA+k-means”。我们对读取计数矩阵进行预处理,然后使用预处理数据作为SIMLR、PCA+k-means和MPSSC的输入。首先,读取计数矩阵按库大小进行标准化,因此各单元的总数是相同的。接下来,对规范化读计数进行对数转换。SIMLR是一种光谱聚类方法,通过多核学习细胞之间的相似性。SIMLR被设置为使用默认设置。MPSSC是一种多核谱聚类框架,在目标矩阵上施加稀疏结构。MPSSC的参数为rho=0.2, lam=0.0001, lam2=0.0001, eta=1, c=0.1。PCA+k-means是利用PCA将处理后的原始读计数矩阵直接投影到二维空间,然后进行k-means聚类的方法。我们遵循作者为CIDR (https://github.com/VCCRI/CIDR)所描述的步骤。CIDR的输入是由原始计数矩阵构造的scData R对象。CIDR的聚类步骤包括确定dropout事件和imputation加权阈值、计算CIDR不相似矩阵、降维和聚类。我们使用CIDR计算的前两个主成分来显示潜在表示。scvis是一种基于变分自编码器50的模型,用于捕获scRNA-seq数据的低维表示。我们使用scvis将scRNA-seq数据减少到二维空间,然后应用k-means聚类。对于scvis,我们遵循作者描述的预处理步骤:每个基因的表达被量化为log2(CPM/10+1),其中“CPM”代表“每百万计数”。然后,通过PCA将数据投影到100维空间,作为scvis的输入。DEC (https://github.com/XifengGuo/DEC-keras)使用与scDeepCluster相同的输入:原始计数矩阵是库大小的归一化,对数转换,缩放和集中。DEC中的超参数与作者的原始参数保持一致(例如,隐藏层的大小为500,500,2000,10)。
Evaluation metrics.
数据模拟。
模拟数据由Splatter R 包生成。利用R函数Splatter 模拟模拟scRNA-seq计数数据。在所有设置中,我们模拟了三个细胞组,2500个基因中的1500个细胞,每个设置重复生成20个数据集。
为了模拟各种丢失率,我们设置了参数丢失率。
shape=−1, vary dropout.mid from −0.5 to 1(−0.5,0,0.5,1;相应的辍学率分别为12±0.3%、17±0.4%、23±0.5%、30±0.6%、39±0.6%,均为默认辍学率。mid in Splatter =0), de.fracScale=0.3;其他参数保持默认值。每个设置生成20个具有不同随机种子的数据集。在生成读计数矩阵之后,Splatter实现一个逻辑函数来产生一个计数应该为零的概率。dropout前的读计数矩阵为真实计数,dropout后的读计数矩阵为原始计数,作为输入进行聚类。由于随机性,具有相同设置的数据集具有略有不同的辍学率。我们总结了每个设置的辍学率的平均值和标准差。计算辍学率的公式定义为
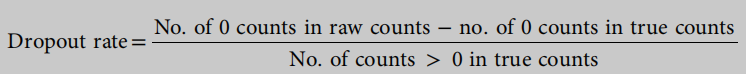
为了模拟具有不同信号强度的数据集,我们改变了参数de。
fracScale在0.2,0.225,0.25,0.275和固定dropout。形状=−1,dropout.mid = 0;其他参数保持默认值。在Splatter中,参数de.fracScale是控制乘法微分表达式因子的对数正态分布的sigma参数。不同de.fracScale设置的效果如图4所示。
我们模拟了一个由3000个基因组成的100000个细胞的大数据集来评估scDeepCluster的运行时间。这10万个细胞被分成10组。参数为dropout。Shape =−1,我们改变dropout。= 0,中期de.fracScale = 0.3。我们将模拟的大型数据集降采样到不同的单元格号码:5,000,7,500,10,000,25,000,50,000,75,000和100,000。降采样没有遗漏任何组。


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









