







一:什么是TCGA数据?TCGA数据有什么作用?
癌症基因组图谱(The Cancer Genome Atlas),简称为TCGA。TCGA数据库是目前最综合全面的癌症病人相关组学数据库。它旨在应用高通量的基因组分析技术,帮助人们对癌症有个更好的认知,从而提高对于癌症的预防、诊断和治疗能力。
二:什么是cgdsr包?如何下载和使用?
R语言工具包,可以下载TCGA数据。下载语句:install.packages("cgdsr")。使用时加载包的语句:library("cgdsr")。
注:R语言中下载包的通用语句为install.packages(需要下载的包)。需要用到包的通用语句为library(需要加载的包)。
三:cgdsr包的初步使用
#load cgdsr packages
library(cgdsr)
#创建一个cgdsr对象(Create a cgdsr object)
mycgds <- CGDS("http://www.cbioportal.org/")
#获取cgdsr对象中存在的数据集,即所有可以研究的癌症类型名的集合(Gets the dataset that exists in the cgdsr object)
all_TCGA_studies <- getCancerStudies(mycgds)
#View the datasets
print(all_TCGA_studies)部分运行结果如下图所示:

从结果图中观察到数据集应该有多列,但是由于数据集过多,而列的空间又有限,结果不方便观察。为了便于观察,我们可以使用R语言中的DT包进行数据的展示。
四:什么是DT包?DT包有什么作用?
data.table包,简称DT包,是R语言中的数据可视化工具包。DT包可以将Javascript中的方法运用到R中,也能将矩阵或者数据表在网页中可视化为表格,以及其它的一些功能。下载和使用的方法与cgdsr包相同。
五:DT的初步使用
#load DT package
library(DT)
#以数据表的形式显示数据集(Present the data set in the form of a data table)
DT::datatable(all_TCGA_studies)部分结果如下图所示。
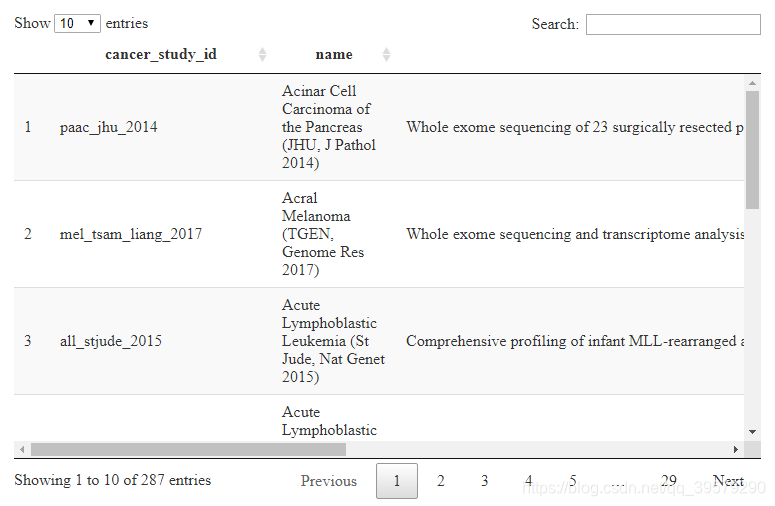
相比直接输入结果,我们可以观察到以表格形式展示出来更便于观察数据及相关信息。从表格中我们可以得出每个数据都包含cancer_study_id,name和description三列信息,分别表示研究癌症的类型,即数据集的名字简称,名字全称和发表的文章题目。
由上图所示,我们可以清晰观察到总共有287种数据集,在实际操作中,可能并不需要研究所有的数据集,所以只需要对特定的数据集进行操作。
六:查看任意数据集的相关信息
(1)查看任意数据集的样本列表方式
#Arbitrarily select a dataset, such as stad_ tcga_ Pub
#研究stad_tcga_pub需要研究的癌症类型名
stad2014 <- "stad_tcga_pub"
#获取特定癌症研究的可用病例列表(Get available case lists for a specific cancer study)
all_tables <- getCaseLists(mycgds, stad2014)
#view how many sample list methods are available in it,the result is two parameters: row and column
dim(all_tables)
DT::datatable(all_tables)结果如下所示:
![]() 即数据集stad_tcga_pub共有11行数据,每行数据有5条相关信息。
即数据集stad_tcga_pub共有11行数据,每行数据有5条相关信息。
如何只需要展示出数据的部分信息,例如只需要所有数据的前三列,可以用以下语句获取相关数据:
DT::datatable(all_tables[,1:3])运行结果如下:

(2)查看任意数据集的数据形式
#获取特定癌症研究的可用基因数据(Get available genetic data profiles for a specific cancer study)
all_dataset <- getGeneticProfiles(mycgds, stad2014)
#以表格形式展示数据
DT::datatable(all_dataset,
extensions = 'FixedColumns',
options = list(
dom = 't',
scrollX = TRUE,
fixedColumns = TRUE
))结果如下所示:
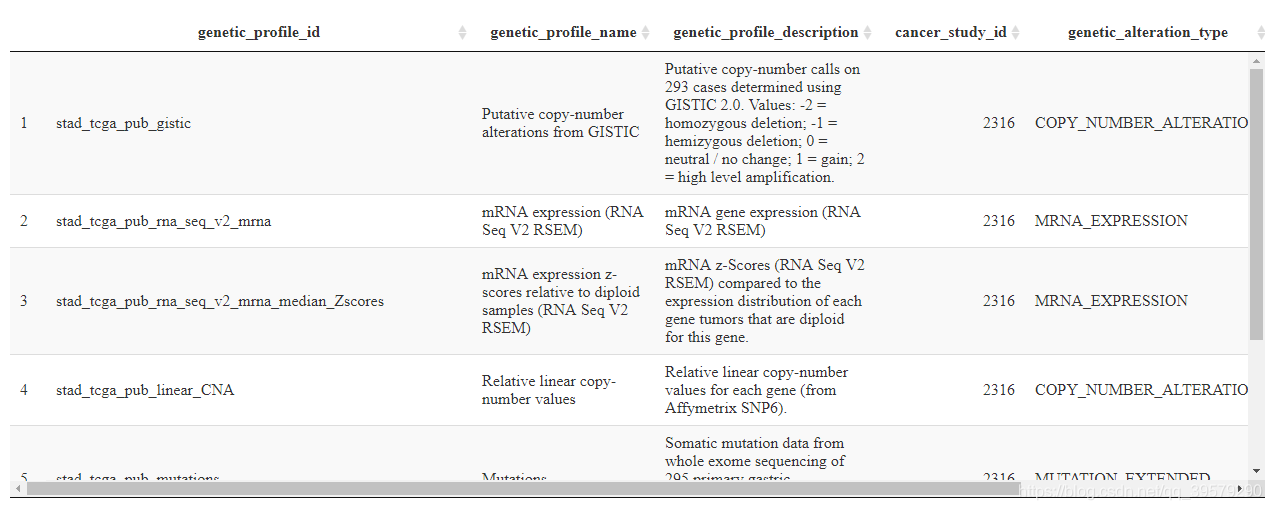
(3)选定数据形式及样本列表后获取感兴趣基因的信息
my_dataset <- 'stad_tcga_pub_rna_seq_v2_mrna'
my_table <- "stad_tcga_pub_rna_seq_v2_mrna"
#检索基因和遗传图谱的基因组图谱数据(Retrieves genomic profile data for genes and genetic profiles)
#getProfileData函数的必要参数为cgdsr object---CGDS对象,genes---基因名称,geneticProfiles---需要研究的病例名称中的可用基因数据,caseList---需要研究的病例名称)
BRCA1 <- getProfileData(mycgds, "BRCA1", my_dataset, my_table)
#获取样本列表
dim(BRCA1)
#以表格形式展示数据
DT::datatable(BRCA1)(4)选定样本列表获取临床信息
#获取特定癌症研究的临床数据(Get clinical data for cancer study)
clinicaldata <- getClinicalData(mycgds, my_table)
#数据展示
DT::datatable(clinicaldata,
extensions = 'FixedColumns',
options = list(
scrollX = TRUE,
fixedColumns = TRUE
))(5)综合性获取(只需要根据癌症列表选择自己感兴趣的研究数据集即可,然后选择好感兴趣的数据形式及对应的样本量。就可以获取对应的信息)
library(cgdsr)
library(DT)
#create a CGDS object
mycgds <- CGDS("http://www.cbioportal.org/")
#指定一个数据集(Specify a dataset)
mycancerstudy = 'brca_tcga'
#获取特定癌症研究的可用病例列表(Get available case lists for a specific cancer study)
getCaseLists(mycgds,mycancerstudy)[,1]
#获取特定癌症研究的可用基因数据(Get available genetic data profiles for a specific cancer study)
getGeneticProfiles(mycgds,mycancerstudy)[,1]
#设置需要研究的病例名
mycaselist ='brca_tcga_rna_seq_v2_mrna'
#设置需要研究的可用基因数据
mygeneticprofile = 'brca_tcga_rna_seq_v2_mrna'
# Get data slices for a specified list of genes, genetic profile and case list
#检索基因和遗传图谱的基因组图谱数据(Retrieves genomic profile data for genes and genetic profiles)
expr=getProfileData(mycgds,c('BRCA1','BRCA2'),mygeneticprofile,mycaselist)
#展示数据
DT::datatable(expr)如下图所示,即为指定基因在指定癌症的表达量。
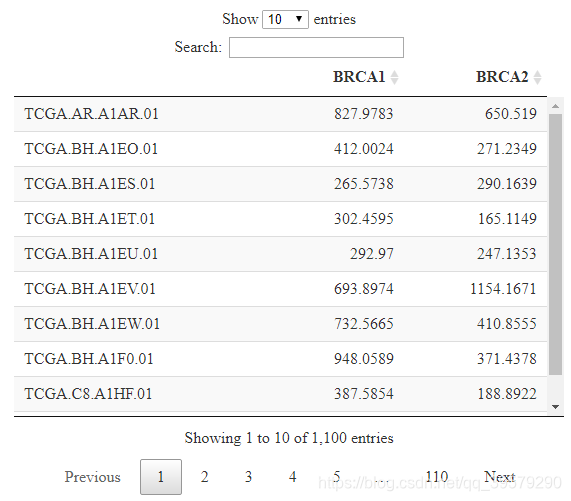
获取临床数据。
#获取病例列表的临床数据(Get clinical data for the case list)
myclinicaldata = getClinicalData(mycgds,mycaselist)
DT::datatable(myclinicaldata,
extensions = 'FixedColumns',
options = list( #dom = 't',
scrollX = TRUE,
fixedColumns = TRUE
))
(6)从cBioPortal下载点突变信息
library(cgdsr)
library(DT)
mycgds <- CGDS("http://www.cbioportal.org/")
#突变基因名称集合
mutGene=c("EGFR", "PTEN", "TP53", "ATRX")
#检索基因和遗传图谱的基因组图谱数据(Retrieves genomic profile data for genes and genetic profiles.)
mut_df <- getProfileData(mycgds,
caseList ="gbm_tcga_sequenced",
geneticProfile = "gbm_tcga_mutations",
genes = mutGene
)
#Apply Functions Over Array Margins
mut_df <- apply(mut_df,2,as.factor)
#数据处理(空值和NAN设置为空,如果不为空设置为MUT)
mut_df[mut_df == "NaN"] = ""
mut_df[is.na(mut_df)] = ""
mut_df[mut_df != ''] = "MUT"
#展示数据
DT::datatable(mut_df)结果如下所示:
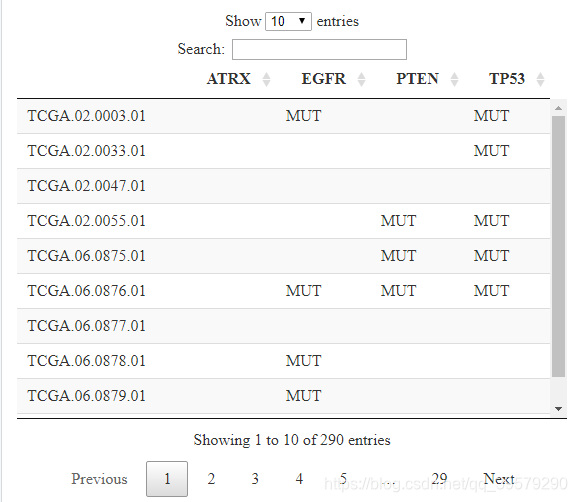
(7)从cBioPortal下载拷贝数变异数据
library(cgdsr)
library(DT)
mycgds <- CGDS("http://www.cbioportal.org/")
mutGene=c("EGFR","PTEN","TP53","ATRX")
cna <- getProfileData(mycgds,
caseList = "gbm_tcga_sequenced",
geneticProfiles = "gbm_tcga_gistic",
genes = mutGene)
cna <- apply(cna,2,function(x)as.character(factor(x,levels=c(-2:2),
labels = c("HOMDEL","HETLOSS","DIPLOID","GAIN","AMP"))))
cna[is.na(cna)] = ""
cna[cna=='DIPLOID']=""
DT::datatable(cna)结果如下所示:

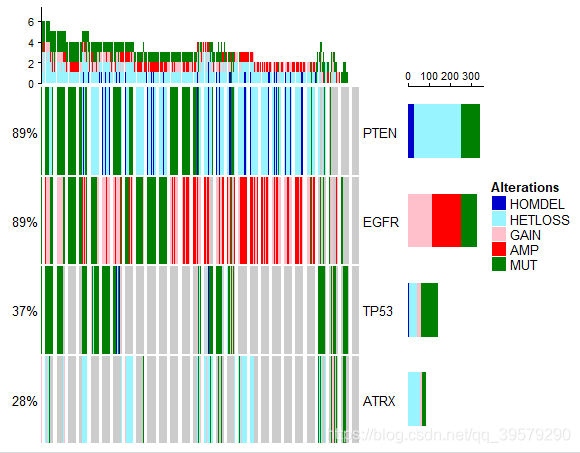
(8)把拷贝数及点突变信息结合画热图
library(grid)
library(ComplexHeatmap)
library(cgdsr)
conb <- data.frame(matrix(paste(as.matrix(cna),as.matrix(mut_df),sep = ";"),
nrow=nrow(cna),ncol=ncol(cna),
dimnames=list(row.names(mut_df),colnames(cna))))
mat <- as.matrix(t(conb))
DT::datatable((mat))
alt <- apply(mat,1,function(x)strsplit(x,";"))
alt <- unique(unlist(alt))
alt <- alt[which(alt !="")]
alt <-c("background",alt)
alter_fun = list(
background = function(x,y,w,h){
grid.rect(x,y,w-unit(0.5,"mm"),h-unit(0.5,"mm"),
gp=gpar(fill="#CCCCCC",col=NA))
},
HOMDEL = function(x,y,w,h){
grid.rect(x,y,w-unit(0.5,"mm"),h-unit(0.5,"mm"),
gp=gpar(fill="blue3",col=NA))
},
HETLOSS = function(x,y,w,h){
grid.rect(x,y,w-unit(0.5,"mm"),h-unit(0.5,"mm"),
gp=gpar(fill="cadetblue1",col=NA))
},
GAIN = function(x,y,w,h){
grid.rect(x,y,w-unit(0.5,"mm"),h-unit(0.5,"mm"),
gp=gpar(fill="pink",col=NA))
},
AMP = function(x,y,w,h){
grid.rect(x,y,w-unit(0.5,"mm"),h-unit(0.5,"mm"),
gp=gpar(fill="red",col=NA))
},
MUT = function(x,y,w,h){
grid.rect(x,y,w-unit(0.5,"mm"),h-unit(0.5,"mm"),
gp=gpar(fill="#008000",col=NA))
}
)
col <- c("MUT"="#008000","AMP"="red","HOMDEL"="blue3",
"HETLOSS"="cadetblue1","GAIN"="pink")
alt = intersect(names(alter_fun),alt)
alt_fun_list <- alter_fun[alt]
col <- col[alt]
oncoPrint(mat=mat,alter_fun = alt_fun_list,
get_type = function(x) strsplit(x,";")[[1]],
col = col)效果图如下所示:

















 1298
1298

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









