






1. 什么是Aviary
Aviary是一个集合多种工具模块的宏基因组分箱工具,具体所集成的模块如图:
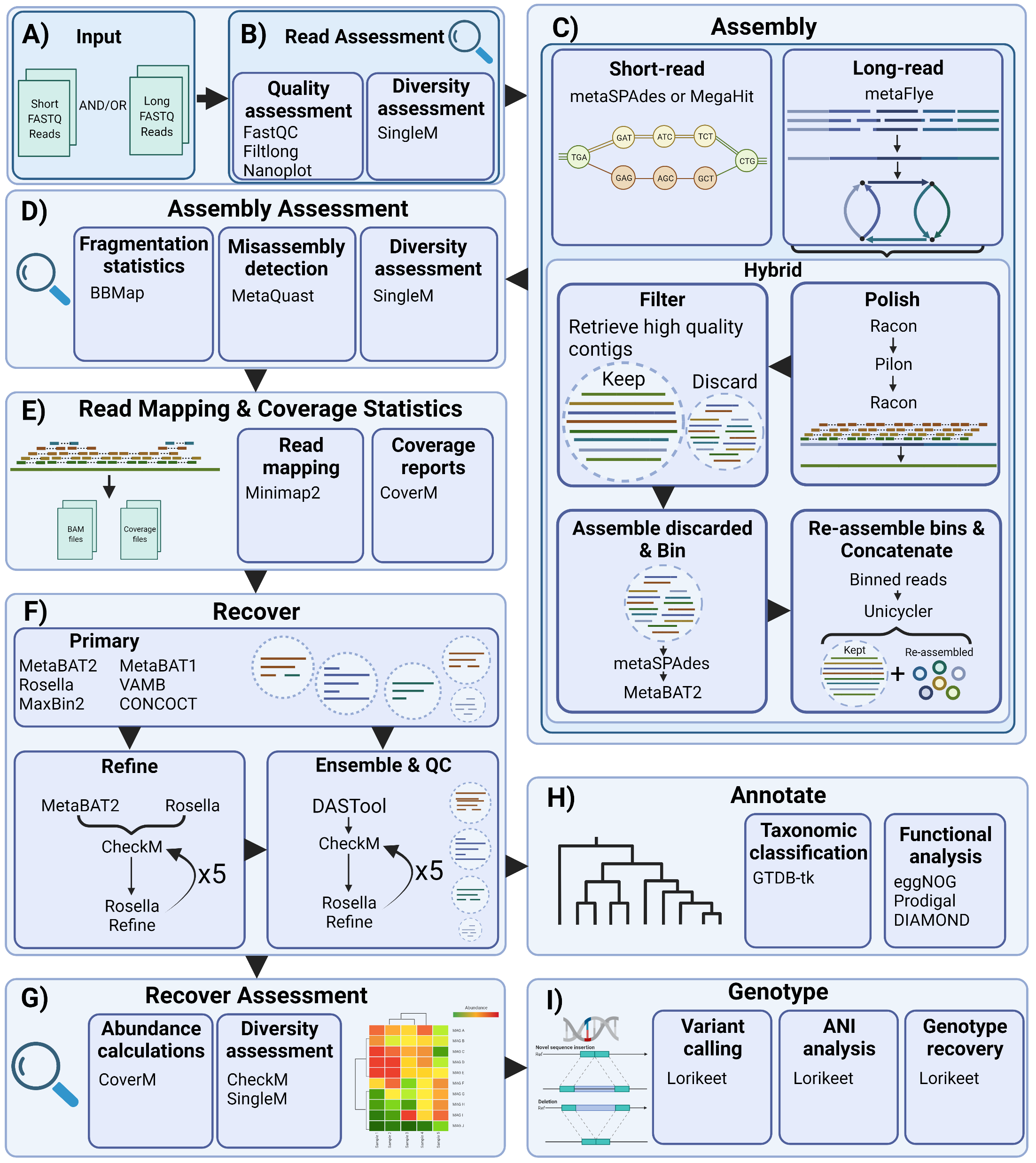
下载安装github: https://github.com/rhysnewell/aviary
2. Recover功能:
其实自己最常使用的就是recover功能了,相较于metawarp对齐速度和refine速度相对较快,但是官方的教程中有点简单,记录一下:
可以直接使用aviary recover -h调出帮助文件
参数分类简解
输入数据相关
-
-1/-2→ 双端测序的 forward/reverse 短读段(Illumina FASTQ)。 -
-i→ interleaved 格式短读段。 -
-c→ coupled 格式的短读段文件。 -
-l→ 长读段(Nanopore 或 PacBio FASTQ)。 -
-z→ 长读段类型(ont=Nanopore, ont_hq=高质量Nanopore, ccs/hifi=PacBio HiFi 等)。 -
-a→ 已有的组装结果(FASTA),如果你已经自己跑过组装,可以直接输入。
组装相关
-
--coassemble→ 是否对多个样本一起组装。 -
--use-megahit→ 短读段组装时用 megahit。 -
--use-unicycler→ 用 Unicycler 来改善混合组装(不推荐复杂宏基因组)。 -
--kmer-sizes→ 手动指定 k-mer 大小(默认 auto)。
质量控制与过滤
-
--min-read-size/--min-mean-q→ 长读段最小长度和平均质量阈值。 -
--min-short-read-length/--max-short-read-length→ 短读段长度过滤。 -
--disable-adpater-trimming→ 是否禁用adapter剪切。 -
--skip-qc→ 跳过 QC。
contig 过滤策略
-
--min-cov-long/--min-cov-short→ 根据覆盖度判断 contig 保留。 -
--exclude-contig-size/--exclude-contig-cov→ 去除低覆盖度、过短的 contig。 -
--include-contig-size→ 强制保留长度超过某值的 contig。 -
-s→ 进入 binning 的 contig 最小长度(默认 1500 bp)。 -
-b→ bin 的最小大小(默认 200 kb)。
binning 相关
-
默认会调用 rosella, semibin, metabat1/2, vamb。
-
--extra-binners→ 额外运行 maxbin、concoct 等。 -
--skip-binners→ 跳过部分 binner。 -
--semibin-model→ SemiBin 的环境模型(human_gut, soil, ocean...)。
后处理与评估
-
--checkm2-db-path→ CheckM2 数据库路径(你解压的 checkm2_database.tar.gz 就是给这个用的)。 -
--gtdb-path→ GTDB 数据库路径。 -
--eggnog-db-path→ EggNOG 数据库路径。 -
--singlem-metapackage-path→ SingleM metapackage 路径。 -
--skip-taxonomy / --skip-singlem / --skip-abundances→ 跳过部分后处理。
计算资源
-
-t→ 每个进程最大线程数。 -
-n→ 最大可用 CPU 核心数。 -
-m→ 最大内存(GB)。 -
--request-gpu→ 是否请求 GPU。 -
--tmpdir→ 临时目录。
Snakemake 工作流控制
-
-w→ 运行的工作流(默认 recover_mags)。 -
--snakemake-profile→ 集群提交配置。 -
--dry-run→ 测试流程,不真正执行。 -
--clean→ 清理中间文件。 -
--build→ 只构建 conda 环境。 -
--download→ 下载数据库(gtdb、eggnog、checkm2…)


















 241
241

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









