







小罗碎碎念
今日推文主题:人工智能在肾脏肿瘤领域的最新研究进展
第一篇文献的关注点在于免疫检查点阻断治疗,我们要学会拆分模型,其实这篇研究就是肾细胞癌+ICB,那么只要我们掌握了临床方面提供的数据是如何用上的,是不是就可以做替换了。
第二篇文章又是单细胞又是空间转录组的,还加上了一个领域泛化原理,标题就看着挺唬人的。小罗的理解是,这个模型在透明细胞肾癌错了测试,效果很好,那么如果有从事相关研究的老师/同学就可以拿过来测试一下。另外,作者说他们提出的模型可以快速识别超过50万个细胞,如果真的效果这么好,那么他们解决过拟合的方法也许你们就可以借鉴了——Dropout。
第三篇文章的思路略微有些反常规,一般都是把RNA测序的数据拿来和切片一起做多模态模型,这里则是不同模态之间的虚拟生成,我觉得数据量很大,且流程足够规范的时候,理论上是可以实现的。
时间有限,我再提一嘴第六篇文献。我其实一直都有在关注癌症的早筛,第六篇文献给我上了一课,我觉得自己的知识面还有非常大的进步空间,纳米阵列和代谢指纹我之前都没听过,一起学习,一起进步吧!!

一、在晚期透明细胞肾细胞癌(aRCC)患者中,如何确定哪些患者可能从免疫检查点阻断(ICB)治疗中获益

一作&通讯
| 角色 | 姓名 | 单位名称(中文) |
|---|---|---|
| 第一作者 | Lisa Kinget | 比利时鲁汶大学实验肿瘤学实验室 |
| 通讯作者 | Benoit Beuselinck | 比利时鲁汶大学大学医院鲁汶癌症研究所医学肿瘤科 |
| 通讯作者 | Abhishek D. Garg | 比利时鲁汶大学细胞应激与免疫实验室 |
文献概述
这篇文章的主要焦点是解决在晚期透明细胞肾细胞癌(aRCC)患者中,如何确定哪些患者可能从免疫检查点阻断(ICB)治疗中获益的问题。
研究团队进行了一项全面的多组学映射分析,涉及超过1000名患者的肿瘤组织,目的是在ICB治疗的背景下理解aRCC的免疫微环境。
研究的关键发现和方法包括:
-
多组学分析:研究者通过对aRCC患者的肿瘤组织进行多组学分析,发现了与免疫反应相关的特定基因和蛋白质的表达模式。
-
HLA分子的特异性:研究指出,具有对肿瘤新抗原具有高特异性的人类白细胞抗原(HLA)等位基因的患者,在接受ICB治疗后,其临床反应更为积极。
-
机器学习模型:研究者开发了一个基于机器学习的模型,用于从肿瘤的转录组数据中推导出一个新的HLA分子的表达特征,这个特征与ICB治疗后的阳性结果相关。
-
小鼠模型实验:使用RENCA肿瘤小鼠模型,研究者发现CD40激动剂与PD1阻断的联合使用可以增强肿瘤相关巨噬细胞和CD8+ T细胞的活性,从而实现最大的抗肿瘤效果。
-
空间映射技术:研究还利用空间转录组技术,对肿瘤微环境中的细胞相互作用进行了空间层面的分析,进一步证实了上述发现。
-
临床验证:研究结果在独立的临床队列中得到了验证,表明这些发现具有潜在的临床应用价值。
-
挑战与限制:尽管ICB治疗在aRCC中已获批准,但目前还没有公认的生物标志物能够合理地预先选择患者或指导“智能”免疫治疗组合。
-
研究意义:这项研究提供了新的多组学和空间地图,揭示了驱动ICB反应的免疫社区结构,有助于改善aRCC患者的临床管理。
文章的详细内容还包括了研究设计、实验方法、数据分析、以及对结果的深入讨论。研究结果不仅增进了对aRCC免疫微环境的理解,而且为开发新的生物标志物和治疗策略提供了科学依据。
重点关注
Fig. 1 提供了该研究的概览和Leuven RWD(真实世界数据)队列的分析结果。
a. 研究设计示意图:这部分描述了研究的发现阶段,研究者首先在aRCC(晚期透明细胞肾细胞癌)的Leuven RWD队列中进行了研究。随后,使用机器学习(ML)模型开发了一个特征标记(signature),并将其在外部(bulk)数据集以及单细胞和空间层面上进行了验证。此外,还通过小鼠RCC/RENCA模型进行了体内功能性验证。

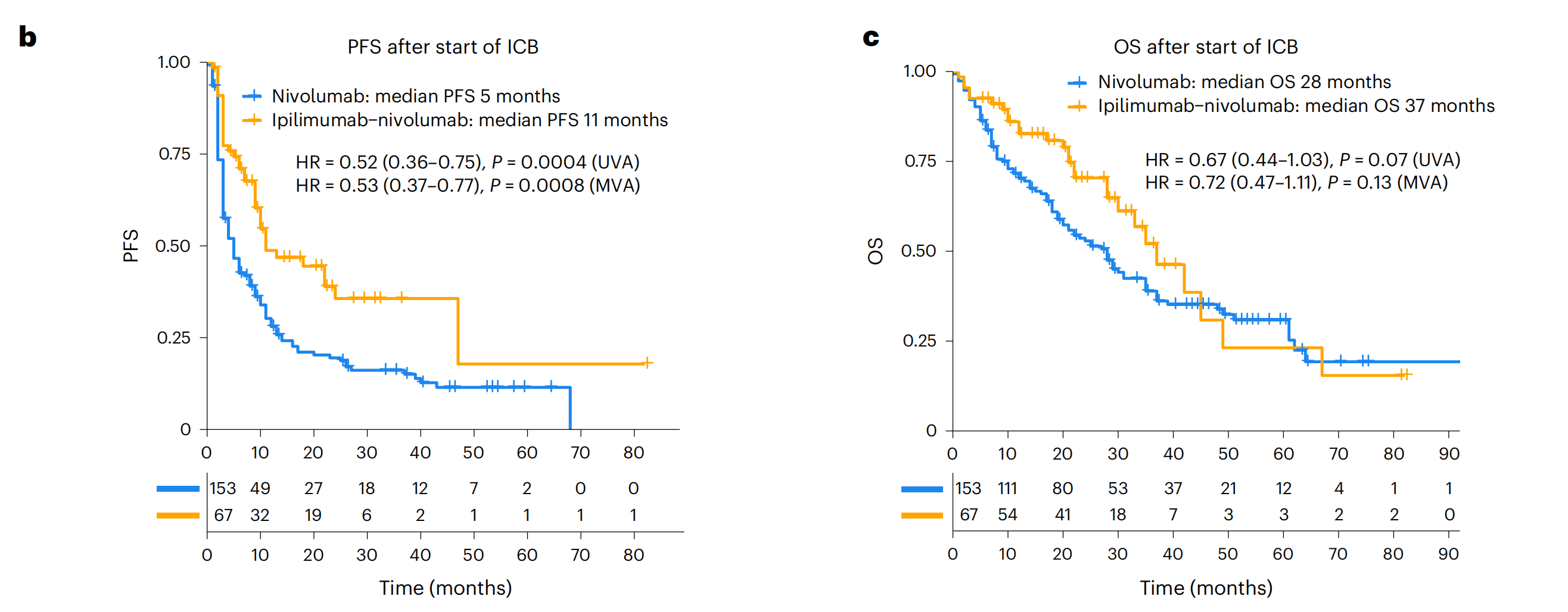
b. PFS的Kaplan-Meier曲线:这部分展示了Leuven RWD队列中,从开始接受ICB(免疫检查点阻断)治疗起,患者的无进展生存期(PFS)。根据ICB治疗的类型(ipilimumab-nivolumab组合治疗与单独nivolumab治疗)进行了分层。使用Cox比例风险回归模型计算了风险比(HR)和置信区间(CI),以比较两种治疗方案的效果。
c. OS的Kaplan-Meier曲线:与b部分类似,这部分展示了总生存期(OS)的Kaplan-Meier曲线,同样根据ICB治疗类型进行了分层,并使用Cox模型计算了HR和CI。
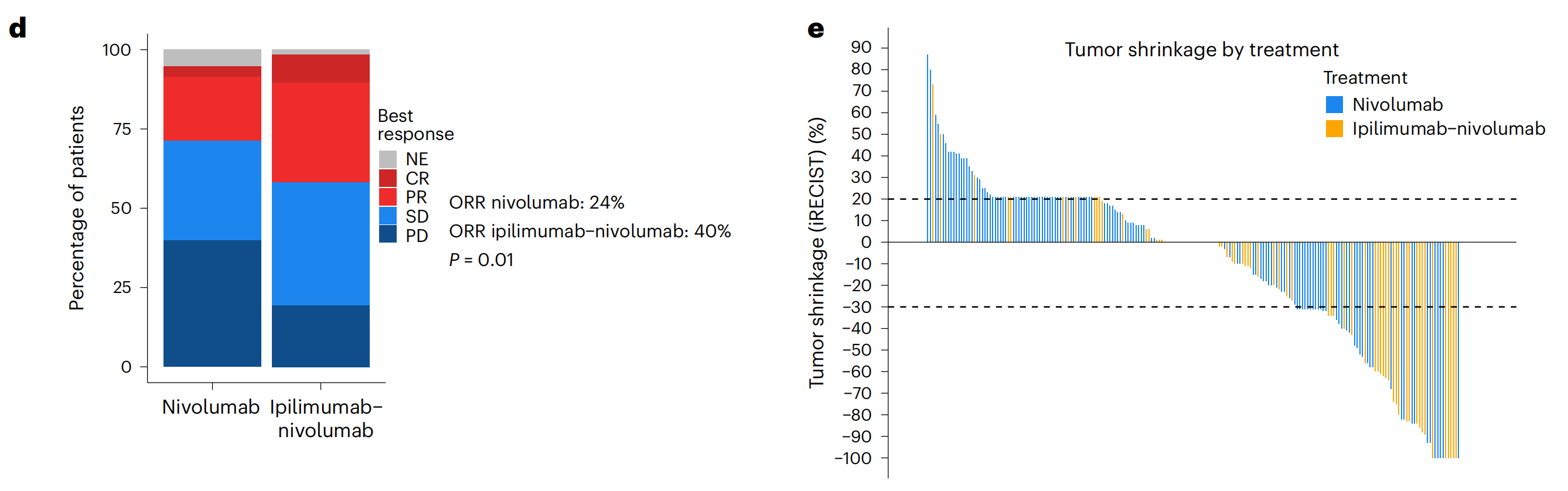
d. 最佳反应的堆叠条形图:这部分通过iRECIST标准(一种评估免疫治疗响应的标准),展示了ICB治疗后患者的最佳反应,包括完全缓解(CR)、部分缓解(PR)、疾病稳定(SD)、疾病进展(PD)和不可评估(NE)。使用Fisher精确检验计算了两种治疗方案间反应差异的P值。
e. 瀑布图:这部分通过iRECIST标准展示了每位患者肿瘤缩小的最大百分比。瀑布图是一种常用于展示肿瘤治疗反应的图表,可以直观地看出不同患者的治疗反应差异。
整体来看,Fig. 1 提供了关于ICB治疗在晚期透明细胞肾细胞癌患者中应用的全面分析,包括了研究设计、临床结果(PFS和OS)、治疗反应评估以及肿瘤缩小情况。通过这些数据,研究者能够评估不同ICB治疗方案的临床效果,并为未来的治疗策略提供科学依据。
二、Cancer-Finder:基于领域泛化原理,高效地在单细胞转录组测序和空间转录组学数据中识别恶性细胞

一作&通讯
| 作者类型 | 作者姓名 | 单位(中文) |
|---|---|---|
| 第一作者 | 钟志兴 | 厦门大学化学化工学院化学生物学系 |
| 第一作者 | 侯俊晨 | 厦门大学生命科学学院药学科学系 |
| 第一作者 | 姚志贤 | 厦门大学化学化工学院化学生物学系 |
| 第一作者 | 董雷 | 上海交通大学医学院瑞金医院病理科 |
| 通讯作者 | 宋佳 | 上海交通大学医学院仁济医院泌尿外科 |
文献概述
这篇文章介绍了一种名为Cancer-Finder的新型深度学习算法,它基于领域泛化(domain generalization)原理,能够高效地在单细胞转录组测序(single-cell RNA sequencing, scRNA-seq)和空间转录组学(spatial transcriptomics, ST)数据中识别恶性细胞。
Cancer-Finder通过从多个具有不同分布的数据集中学习,构建了一个能够泛化到未知领域的模型,从而在单细胞数据中直接区分恶性和正常细胞。该算法在识别恶性细胞方面表现出95.16%的平均准确率,并且能够扩展到空间转录组数据中,准确识别肿瘤组织中的恶性区域。
Cancer-Finder的主要特点包括:
- 利用领域泛化技术,提高算法在未知数据集上的泛化能力。
- 采用深度神经网络,包含特征提取和分类模块,通过随机dropout层防止过拟合。
- 应用风险外推方法,最小化跨领域的风险差异,提高模型在所有组织中的准确性。
- 通过集成解释性模块,识别区分恶性和非恶性细胞的关键特征。
- 在多组织类型的单细胞数据集上进行训练和验证,表现出高准确性和稳健性。
研究者们还使用Cancer-Finder在5个透明细胞肾癌(clear cell renal cell carcinoma, ccRCC)的空间转录组样本上进行了测试,成功识别了肿瘤-正常组织界面处显著共定位并富含的10个基因,这些基因与透明细胞肾癌患者的预后密切相关。
此外,Cancer-Finder在处理大型数据库时展现出显著的速度优势,能够快速预测超过50万个细胞。在与其他现有工具的比较中,Cancer-Finder在预测准确性、速度和内存消耗方面均表现出色。
最后,Cancer-Finder在分析ccRCC的空间异质性方面也显示出潜力,能够提供有关肿瘤微环境(tumor microenvironment, TME)的重要见解,并有助于发现与预后相关的生物标志物。
重点关注
Fig. 1 展示了 Cancer-Finder 的整体架构及其应用。
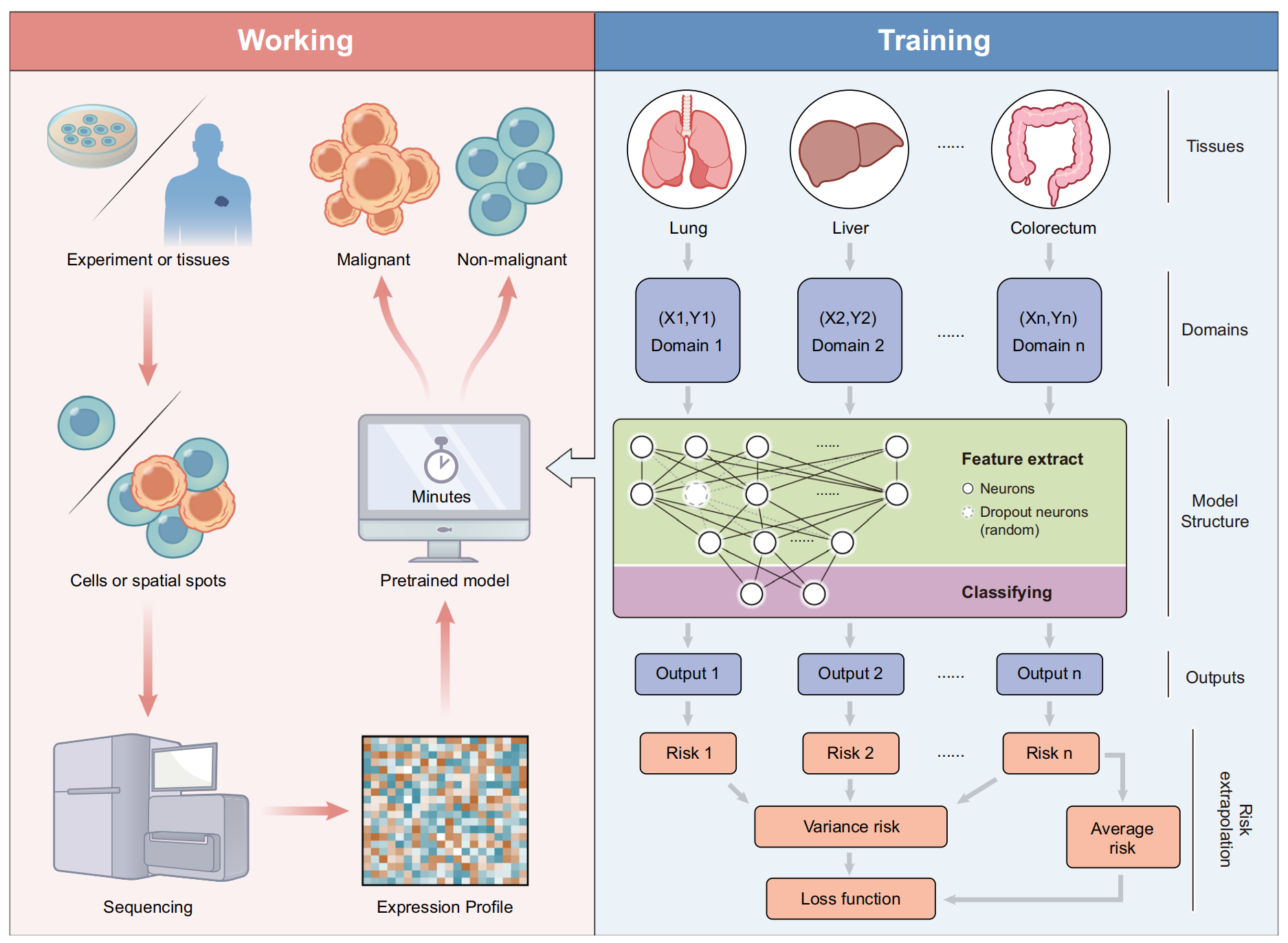
Cancer-Finder 是一个可扩展的框架,它利用单细胞测序数据来准确注释细胞的恶性状态,并且可以轻松扩展到其他数据类型(例如空间转录组数据)。预训练模型能够快速准确地识别来自癌组织的恶性细胞。
为了对抗不同组织之间的差异,Cancer-Finder 采用了领域泛化训练策略,以提高模型在未知领域的普遍区分性能,并准确识别恶性细胞。在这里,“领域”指的是模型训练时使用的具体数据类型或类别。研究者将这一概念应用于单细胞或空间数据中细胞恶性状态的注释,假设来自不同组织的数据显示出不同的领域特征。
Cancer-Finder 的模型是一个包含输入层、两个隐藏层(用于特征提取)以及一个用于分类的层的神经网络。为了实现领域泛化,Cancer-Finder 使用了风险外推方法。这种方法通过减少跨训练领域的风险差异,优化了模型在所有组织中的高准确率,因为降低风险差异可以减少模型对广泛分布变化的敏感性。
为了评估模型在多个领域中的表现,Cancer-Finder 最小化了两种类型的全局风险:方差风险和平均风险。方差风险反映了不同训练领域中风险的离散程度,而平均风险则反映了跨所有训练领域的总体风险。通过最小化这两种风险,Cancer-Finder 能够在不同的数据领域中实现更好的泛化性能。
三、RNA-CDM:利用人类肿瘤的RNA测序数据生成合成的全切片图像瓦片

一作&通讯
| 作者类型 | 姓名 | 单位名称(中文) |
|---|---|---|
| 第一作者 | Francisco Carrillo-Perez | 斯坦福大学医学院生物医学信息学研究中心 |
| 通讯作者 | Olivier Gevaert | 斯坦福大学医学院生物医学数据科学系 |
文献概述
这篇文章讨论了一种名为RNA-CDM的新型机器学习模型,该模型能够利用人类肿瘤的RNA测序数据生成合成的全切片图像瓦片。
这种方法可以解决在获取多样化和足够大的数据集时面临的成本高昂和挑战性问题,特别是在数据稀缺的情况下,通过预训练机器学习模型和数据插补来加速机器学习模型的开发。
研究者们展示了RNA-CDM模型如何通过级联扩散模型从RNA测序数据的潜在表示中合成现实感强的全切片图像瓦片。这些合成图像瓦片能够准确保留细胞类型的分布,并维持在批量RNA测序数据中观察到的细胞比例,这在肺癌、肾乳头状细胞癌、宫颈癌、结肠癌和胶质母细胞瘤等多种癌症类型中都得到了证明。
文章还讨论了癌症的多尺度和多因素特性,以及如何通过不同的数据模态(如RNA测序、全切片成像、微小RNA测序或DNA甲基化)来创建有前景的临床决策支持系统。尽管如此,深度学习模型通常需要大量的数据进行适当的训练,而且大多数现有的数据集是不完整的,缺少某些模态。为了解决这些问题,研究者们提出了生成模型作为解决方案,特别是生成对抗网络(GANs)和变分自编码器(VAE)被用于生成合成的全切片成像和RNA测序数据。
RNA-CDM模型通过一个称为Beta-VAE的模型将RNA测序数据投影到低维潜在空间中,然后使用这个降维表示来生成图像。该模型能够根据不同癌症类型的RNA测序数据生成图像瓦片,并且在没有明确标签信息的情况下,准确合成了五种不同癌症类型的图像瓦片。
研究者们还使用最先进的细胞分割模型HoverNet对真实和合成的图像瓦片进行了评估,发现合成瓦片中的细胞分布与真实数据相似,表明RNA-CDM能够生成具有现实细胞形态的样本。此外,通过使用来自不同癌症类型的RNA测序数据,研究者们证明了合成数据可以用于预训练模型,从而在生物医学分类任务上提高性能。
最后,文章讨论了RNA-CDM模型的潜在应用,包括作为数据增强策略,以及在创建多模态机器学习模型方面的潜力。尽管如此,文章也指出了一些挑战和未来工作的方向,例如如何利用空间转录组学技术生成局部真实的RNA表达配置文件,以及如何处理来自不同队列的RNA表达数据的潜在变异性。
重点关注
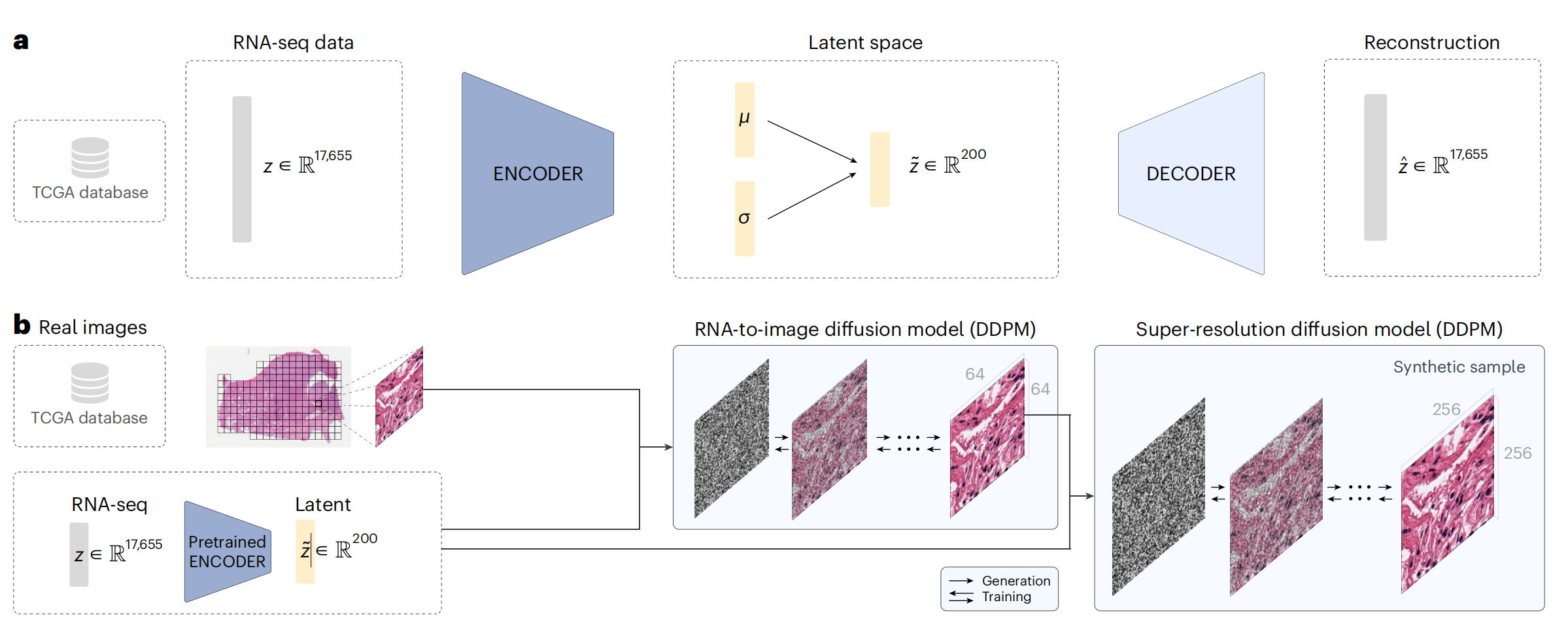
图1展示了RNA-CDM(RNA-to-image Cascaded Diffusion Model)模型架构,该模型用于从RNA测序数据生成合成的全切片图像瓦片。
模型架构分为两个主要部分:
a. Beta-VAE架构:用于生成基因表达嵌入。这个变分自编码器(VAE)模型接收17,655个基因的表达数据作为输入。编码器和解码器均由两个线性层组成,分别包含6,000和4,000个神经元。模型通过这两个层来学习数据的潜在表示,即μ(平均)和σ(标准差)向量,这些向量构成了一个200维的特征空间,用于捕捉基因表达数据的主要特征。
b. RNA-CDM架构:由两个去噪扩散概率模型(Denoising Diffusion Probabilistic Models, DDPM)组成,用于生成合成的多癌症类型图像瓦片。第一个DDPM作为RNA到图像的转换模型,第二个DDPM作为超分辨率模型。
具体步骤如下:
- 使用预训练的编码器从RNA-seq数据中获取潜在表示。
- 从患者那里获取相应的瓦片图像。
- 对于第一个DDPM,将256×256像素的瓦片图像调整大小至64×64像素。
- 在训练阶段,根据每个时间步t的给定噪声计划逐渐向瓦片添加噪声。
- 第一个DDPM学习如何使用噪声图像、时间步t和基因表达嵌入作为输入来减少噪声。
- 在每个时间步,预测的噪声被从噪声图像中移除,最终得到一个去噪的64×64像素瓦片。
- 第二个DDPM接收去噪后的64×64图像、原始的256×256噪声图像、时间步t和基因表达嵌入,预测要添加的噪声。
- 然后,从256×256像素的瓦片中迭代地移除噪声,直到获得与原始瓦片相比较的去噪图像。
在生成新的图像时,过程与上述相同,但起始于完全随机的噪声,直到生成一个合成瓦片,其生成过程受到基因表达嵌入的指导。
这种架构的优势在于能够从RNA测序数据中学习并生成具有高分辨率和多癌症类型的合成图像瓦片,同时保持细胞类型的分布和细胞比例,这对于机器学习模型的预训练和数据插补非常有用。
四、肾细胞癌(RCC)分子亚型对免疫治疗和靶向治疗结果的影响

一作&通讯
| 作者类型 | 作者姓名 | 单位名称(中文) |
|---|---|---|
| 第一作者 | Rene´ e Maria Saliby | 波士顿达纳-法伯癌症研究所医学肿瘤科 |
| 第一作者 | Chris Labaki | 波士顿贝斯以色列女执事医疗中心内科 |
| 通讯作者 | Toni K. Choueiri | 波士顿达纳-法伯癌症研究所医学肿瘤科 |
| 通讯作者 | David A. Braun | 耶鲁大学分子与细胞肿瘤中心(CMCO) |
文献概述
这篇文章是一篇关于肾细胞癌(RCC)分子亚型对免疫治疗和靶向治疗结果影响的研究。
主要内容包括:
- 免疫检查点抑制剂(ICIs)的出现彻底改变了转移性肾细胞癌(mRCC)的治疗管理,FDA批准了包括ICI组合或ICI与抗血管生成酪氨酸激酶抑制剂(VEGF-TKI)的治疗方案。
- 研究者们在IMmotion151(IM151)试验中对823个肿瘤进行了分子分析,识别出七个转录组定义的亚组,这些亚组影响了治疗反应。其中,4、5和7号亚组在接受阿特珠单抗+贝伐珠单抗治疗时,比接受舒尼替尼治疗有更好的结果。
- 研究者们进一步分析了JAVELIN Renal 101(JR101)试验的患者转录组和临床特征,以评估这些分子亚组是否代表真正不同的生物学状态,并且是否能够预测对两种治疗方案(avelumab+axitinib [ICI+VEGF-TKI] 对比 sunitinib)的不同临床结果。
- 通过机器学习模型对IM151试验的肿瘤转录组数据进行训练,并将JR101试验中的734个肿瘤分类为七个分子亚型。两个试验的基线人口统计数据相似。
- 分子亚组与国际转移性肾细胞癌数据库联盟(IMDC)预后风险组有关联。与IM151数据集一致,JR101中特定分子亚组的肿瘤表现出不同的生物学状态,并且每种分子亚型都有独特的体细胞突变景观。
- 在IM151数据集中,特定分子亚型的患者在ICI+VEGF治疗与VEGF-TKI治疗(舒尼替尼)相比显示出不同的反应和生存结果。然而,在JR101数据集中,所有分子亚型的患者在ICI+VEGF-TKI方案(avelumab+axitinib)治疗下的反应率都比舒尼替尼高。
- 尽管这些分子亚型代表了不同的生物学状态,但在JR101研究中,avelumab+axitinib相比舒尼替尼在所有分子亚型中都倾向于改善临床结果。这表明这些亚型可能尚未直接适用于预测ICI+VEGF-TKI与单独VEGF-TKI的预测生物标志物。
- 研究还讨论了生物标志物在RCC治疗中的复杂性,并强调了需要进一步研究和验证以完善我们对RCC分类和个性化治疗策略的理解。
五、使用基因组规模代谢模型和突变数据预测与癌症体细胞突变相关的代谢物

一作&通讯
| 作者类型 | 姓名 | 单位名称(中文) |
|---|---|---|
| 第一作者 | GaRyoung Lee | 韩国高等科学技术院化学与生物分子工程系 |
| 第一作者 | Sang Mi Lee | 韩国高等科学技术院化学与生物分子工程系 |
| 通讯作者 | Hongseok Yun | 首尔国立大学医院基因组医学科 |
| 通讯作者 | Youngil Koh | 首尔国立大学医院内科 |
| 通讯作者 | Hyun Uk Kim | 韩国高等科学技术院化学与生物分子工程系 |
文献概述
这篇文章的主要研究内容是,使用基因组规模代谢模型和突变数据预测与癌症体细胞突变相关的代谢物。
作者们开发了一个计算工作流程,用于预测与特定癌症体细胞突变显著相关的代谢物-基因-途径集合(MGPs)。这些MGPs有助于识别新的肿瘤代谢物,并可能为癌症治疗策略提供参考。
研究背景指出,肿瘤代谢物通常由基因突变产生,并在癌细胞中积累,从而促进肿瘤发生。然而,由于细胞内代谢物数量庞大,以及与癌症发展相关的多个基因,识别这些与突变相关的代谢物具有挑战性。
研究结果表明,作者们构建了一个计算模型,该模型利用癌症患者的基因组规模代谢模型(GEMs)和突变数据来生成MGPs。GEMs是一种计算模型,可以预测基因组规模的反应通量,并可以使用组学数据以细胞特异性方式构建。该计算工作流程首先通过与急性髓系白血病和肾细胞癌样本的多组学数据(包括突变数据、RNA-seq数据和代谢组数据)比较结果来验证。然后,进一步使用公开可用的RNA-seq数据,对18种癌症类型的预测MGPs进行评估,并与报告的研究进行比较。
文章的结论是,计算工作流程验证表明,大量代谢物和代谢途径似乎与特定的体细胞突变显著相关。该计算工作流程和得到的MGPs将有助于识别新的肿瘤代谢物,并提出癌症治疗策略。
研究使用了来自PCAWG和TCGA的RNA-seq数据,重建了1056个癌症患者特异性的GEMs,并预测了25种癌症类型的MGPs。研究还讨论了这些预测MGPs的治疗潜力,并以CNS-GBM/Oligo的MGPs为例,与先前研究报告的发现进行了比较分析。
文章还讨论了计算工作流程的局限性和未来改进的可能性,指出需要更多的样本和多组学数据来加强MGPs的验证,并系统地分析癌症类型特异性的代谢情况。作者们希望这个计算工作流程能够为发现新的肿瘤代谢物和促进各种治疗和诊断策略的发展奠定基础。
最后,文章提供了使用不同工具和数据库进行数据处理和分析的方法,包括质量控制、序列比对、变体检测、基因表达估算和代谢组数据分析等。
重点关注
图1展示了一个用于预测与癌症相关的代谢物-基因-途径集合(MGPs)的计算工作流程。这个流程利用了癌症患者的基因组规模代谢模型(GEMs),并针对癌症中的突变基因列表,对每个代谢物重复进行分析。
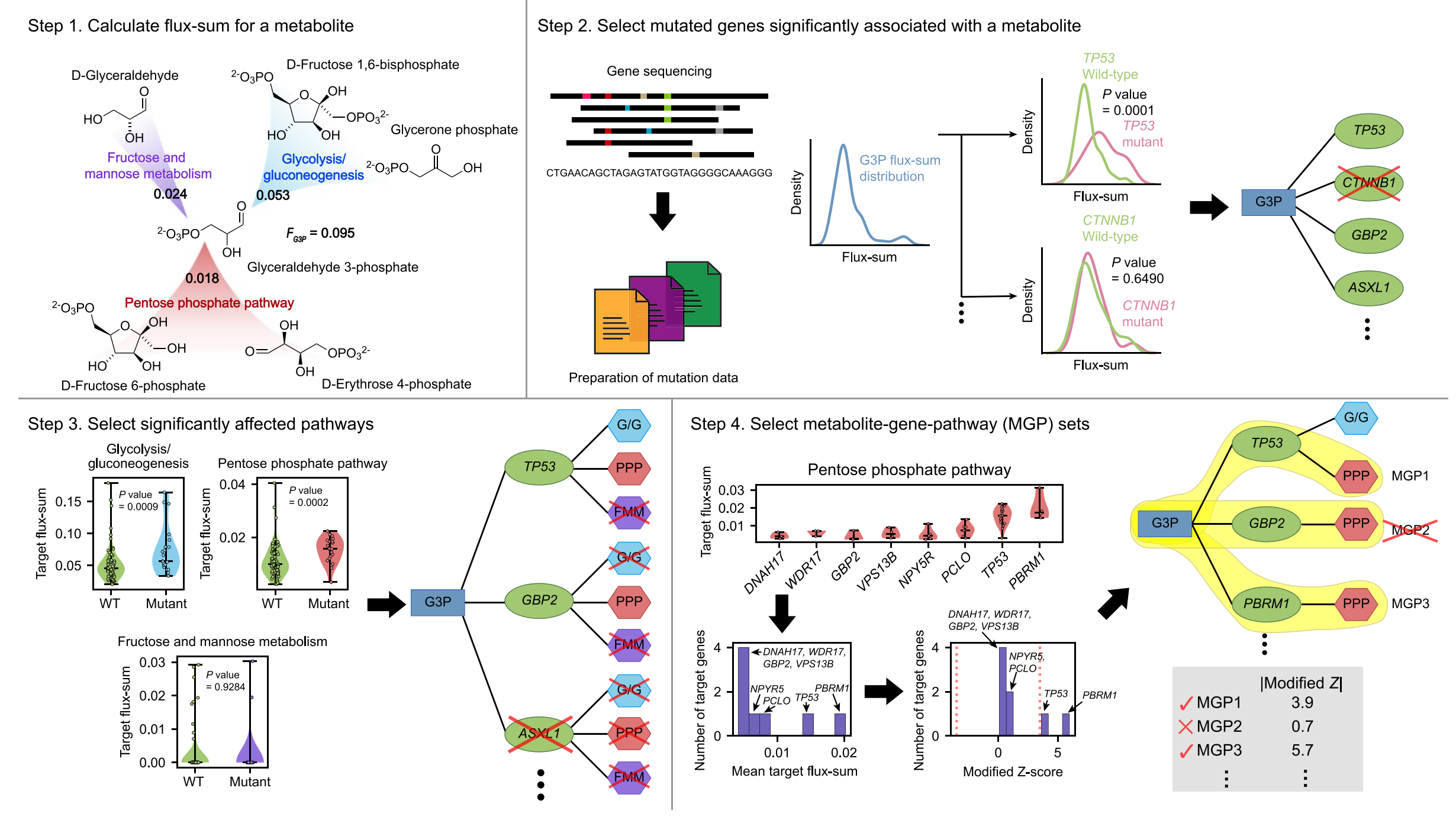
-
使用RNA-seq数据生成患者特异性GEM:首先,基于RNA-seq数据为每位癌症患者构建一个基因组规模的代谢模型(GEM)。这个模型预测了在基因组层面上所有代谢反应的通量。
-
预测代谢物的通量和值(Flux-sum值):对于目标代谢物,使用患者特异性的GEM来预测其通量和值。Flux-sum值是指为了生成或消耗该代谢物所需的所有通量的总和,它在本质上代表了代谢物在稳态条件下的周转率。
-
基因与代谢物配对(MG配对):如果某个基因突变导致代谢物的通量和分布显著不同,那么这个代谢物就与该基因配对。这一步骤识别了可能与特定基因突变相关的代谢物。
-
连接代谢途径:将上一步预测的MG配对与生物合成目标代谢物的代谢途径联系起来。如果这些途径在目标基因突变时显示出显著不同的“目标通量和值”,则认为这些途径与MG配对相关。
-
筛选MGP候选:如果在第4步中没有找到目标途径,则移除相应的MG配对。这一步确保了只有那些与特定代谢途径相关的MG配对才会被保留。
-
选择MGPs:最后,通过识别每个目标途径中那些显示出与同一途径中其他目标基因显著不同的目标通量和值的目标基因来选择MGPs。对于每个目标途径中的目标基因,计算其目标通量和值的平均数,并将其转换为修正的Z分数。只有当MGPs的修正Z分数满足阈值“3.5”时,它们才会被最终选择。
整个工作流程是一个迭代的过程,它从代谢物的通量预测开始,通过一系列的筛选步骤,最终识别出与特定基因突变显著相关的代谢物和代谢途径。这种方法有助于揭示癌症细胞中代谢变化的分子机制,并可能为癌症的治疗提供新的策略和靶点。
六、通过激光解吸/电离质谱(LDI-MS)分析血清和尿液中的代谢指纹,以实现早期肾细胞癌(RCC)的诊断

一作&通讯
| 作者角色 | 姓名 | 单位名称(中文) |
|---|---|---|
| 第一作者 | Yuning Wang | 上海交通大学系统医学研究院、生物医学工程学院、医疗机器人研究院 |
| 第一作者 | Xiaoyu Xu | 上海交通大学系统医学研究院、生物医学工程学院、医疗机器人研究院 |
| 通讯作者 | Wei Zhai | 上海交通大学医学院附属仁济医院泌尿外科 |
| 通讯作者 | Kun Qian | 上海交通大学系统医学研究院、生物医学工程学院、医疗机器人研究院 |
文献概述
这篇文章是关于一种新型自组装超支化金纳米阵列(HyBrAuNA)的研究,该技术用于通过激光解吸/电离质谱(LDI-MS)分析血清和尿液中的代谢指纹,以实现早期肾细胞癌(RCC)的诊断。
研究的核心是开发了一种基于HyBrAuNA的LDI-MS平台,该平台通过自组装技术在油水界面形成紧密排列的纳米阵列,从而显著增强了电离效率和质谱信号的重复性。HyBrAuNA具有强大的电磁场增强和高光热转换效率,能够有效地电离低丰度的代谢物,用于收集血清和尿液的代谢指纹(S-UMFs)。
研究结果表明,该平台与自动化机器学习分析相结合,能够以超过0.99的曲线下面积(AUC)区分早期RCC患者和健康对照组。此外,研究还筛选出一组9种代谢物(4种来自血清,5种来自尿液)及其相关途径,这些代谢物和途径可能对早期肾肿瘤的诊断具有重要意义。
文章强调了该平台的高吞吐量、快速分析速度和低样品消耗的特点,认为其在生物流体的代谢分析、疾病诊断和病理机制探索方面具有潜力。研究还讨论了RCC的流行病学、早期诊断的重要性以及目前临床诊断方法的局限性。此外,文章还详细介绍了HyBrAuNA的制造过程、表征、以及LDI-MS平台的性能评估。
最后,研究团队提出了未来的研究方向,包括在更大的多中心队列中探索HyBrAuNA基于LDI-MS平台的应用价值,以及进一步阐明异常代谢物水平与RCC发展之间的关系,从而为RCC治疗提供指导。
重点关注
Scheme 1 描述了一种基于自组装超支化黑金纳米阵列(HyBrAuNA)的激光解吸/电离质谱(LDI-MS)平台,用于血清和尿液代谢指纹(S-UMFs)的分析,目的是对早期肾细胞癌(RCC)进行诊断。

-
HyBrAuNA的制造流程:首先,使用金种子(Au seeds)作为原始材料,通过连续岛屿生长策略合成超支化黑金纳米粒子(hyperbranched black AuNPs)。在这个过程中,多巴胺自聚合形成聚多巴胺,并在金种子表面原位吸附,同时金前体被聚多巴胺还原,形成具有黑色外观的超支化结构。
-
HyBrAuNA的自组装:超支化黑金纳米粒子通过静电相互作用在油水界面自组装成有序紧密排列的纳米阵列,形成HyBrAuNA。这一步骤涉及到使用带正电荷的硫醇胺(cysteamine)稳定的金纳米粒子,以促进在油水界面的有序排列。
-
HyBrAuNA基LDI-MS平台的性能展示:通过实验和理论研究,展示了HyBrAuNA辅助的LDI-MS性能,包括高灵敏度和出色的重复性,适用于代谢物分析。
-
血清和尿液代谢指纹的提取与分析:将HyBrAuNA基LDI-MS平台应用于血清和尿液样本的代谢分析,以探索早期RCC的代谢变化。通过自动化机器学习(AutoML)分析S-UMFs,能够以高准确度区分早期RCC患者和健康对照组。
-
早期RCC诊断的分类模型构建:使用AutoML技术构建基于S-UMFs的分类模型,通过优化的超参数和分类算法,提高了早期RCC诊断的性能。
-
代谢物标记的筛选:通过AutoML分析,筛选出一组9种代谢物(4种来自血清,5种来自尿液)作为早期肾肿瘤的潜在生物标志物。
Scheme 1 强调了HyBrAuNA基LDI-MS平台的创新性和实用性,展示了其在早期RCC诊断中的潜力,特别是在提高诊断准确性和简化操作流程方面。



















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









