







小罗碎碎念
细胞核分割与分类
在医学AI领域,细胞核分割与分类是数字病理学工作流程的关键环节。
每张全玻片图像(WSI)可能包含多达数百万个不同类型的细胞核,这些细胞核可通过系统分析用于预测临床结果。
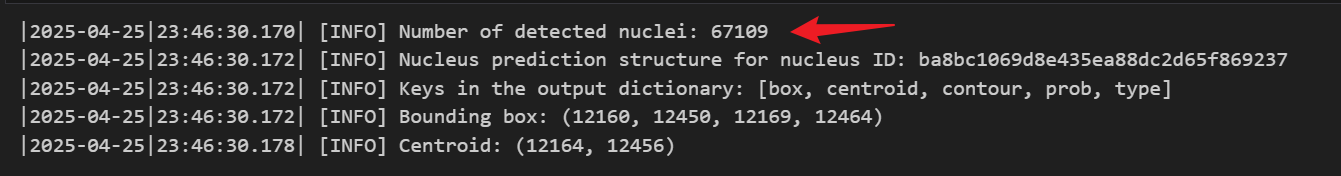
我这里简单展示一下自己的测试结果,切片大小为190M,相对较小,使用的设备为4060,GPU占用率为74%,耗时80min,最后检测出67109个细胞核。
我们前面介绍过很多病理AI领域的顶刊,尤其是结合空间转录组学、基因组学的文章,在解释模型效果以及肿瘤微环境的时候,非常喜欢使用Hovernet。
知识星球和交流群内,也经常有人提到这个模型,并且目前网上没有系统详细的复现教程,所以这一期推送就手把手带着大家解决这个问题。
最终的可视化分析结果主要以两种形式展现,包括:tile级别的推理、Slide级别的推理。
项目复现
在此示例中,我们将演示如何使用TIAToolbox中实现的HoVer-Net来应对这些挑战,并解决组织学图像中的细胞核实例分割与分类问题。
本期项目复现使用的设备是Windows,GPU型号为4060(考虑到大部分粉丝使用的是Windows,所以新购入的设备,哈哈),不过Liunx/Mac用户也适用,因为主要的区别在于环境的配置和路径的指定,我都会在后续提到。
HoVer-Net是一种基于深度学习的方法,其名称源于细胞核像素到对应细胞核质心的水平和垂直距离(HoVer),这些距离用于分离成簇的细胞核。对于每个分割的实例,随后通过专用的上采样分支确定细胞核类型。
我们主要使用NucleusInstanceSegmentor,其默认使用预训练的HoVerNet模型之一。我们还将介绍TIAToolbox中嵌入的可视化工具,用于将实例分割结果叠加到输入图像上。
文献概述
本文提出的HoVer-Net,一直是细胞核分割分类领域的常用模型。

HoVer-Net是一种新型卷积神经网络,它利用核像素到质心的水平和垂直距离信息,实现细胞核的同时分割与分类。
网络架构基于预激活残差网络,通过三个上采样分支分别完成核像素检测、距离预测和核类型分类任务。独特的损失函数设计以及基于Sobel算子的后处理方法,有效提升了模型性能。在多个多组织组织学图像数据集上的实验表明,HoVer-Net相比其他方法,在细胞核实例分割和分类任务中均取得了领先成果。
此外,文章还引入了新的结直肠核分割和表型(CoNSeP)数据集,包含24,319个标注细胞核,为相关研究提供了更丰富的数据支持。
同时,提出了全景质量(PQ)等评估指标,能更准确地衡量模型性能。总体而言,HoVer-Net为医学AI中细胞核相关研究提供了新的思路和方法,具有重要的应用价值 。
阅前必读
注意,由于编写项目复现系列教程需要花费大量时间,所以采取付费阅读的形式,绝对让你物超所值,你通过这一篇推送,可以帮助你节省大量自己摸索的时间。
【1】阅读方式1:知识星球(推荐)
已订阅星球用户可以前往知识星球获取pdf版本教程,并且可以在星球中提问,我会给出详细解答。此外,星球是按年付费,更划算!
【2】阅读方式2:微信推送付费阅读
如果只对单篇内容感兴趣,可以直接支付本篇文章费用。
交流群
欢迎大家加入【医学AI】交流群,本群设立的初衷是提供交流平台,方便大家后续课题合作。
目前小罗全平台关注量67,000+,交流群总成员1500+,大部分来自国内外顶尖院校/医院,期待您的加入!!
由于近期入群推销人员较多,已开启入群验证,扫码添加我的联系方式,备注姓名-单位-科室/专业,即可邀您入群。
知识星球
对推文中的内容感兴趣,想深入探讨?在处理项目时遇到了问题,无人商量?加入小罗的知识星球,寻找科研道路上的伙伴吧!
一、文献概述
文章提出的HoVer-Net,用于多组织组织学图像中细胞核的同时分割与分类,有效解决了传统方法面临的难题,推动了医学图像分析领域的发展。
1-1:研究背景与挑战
手动评估苏木精 - 伊红(H&E)染色组织学切片存在通量低、观察者间和观察者内变异性大的问题。
数字病理学虽能解决部分难题,但细胞核分割和分类面临挑战。细胞核具有高度异质性,不同细胞类型、疾病类型的细胞核在形状、大小和染色质模式上存在显著差异,肿瘤细胞核还常聚集重叠,增加了自动分割的难度;
同时,由于细胞核外观变化大,分类也颇具挑战。
1-2:方法
HoVer-Net基于预激活残差网络(Preact-ResNet50)构建,通过三个上采样分支实现细胞核实例分割与分类。
核像素(NP)分支区分核像素与背景,HoVer分支预测核像素到质心的水平和垂直距离以分离聚集核,核分类(NC)分支确定每个核的类型。
采用多任务损失函数,包括HoVer分支的回归损失、NP和NC分支的交叉熵损失与骰子损失,联合优化网络权重。
后处理阶段,利用HoVer分支输出的水平和垂直图计算梯度,确定细胞核分离位置,通过阈值函数和标记控制的分水岭算法实现实例分割,并将NC分支的像素级预测转换为核实例的类型预测。
1-3:评估指标
提出全景质量(PQ)指标评估细胞核实例分割性能,该指标分解为检测质量(DQ)和分割质量(SQ),能更全面地评估模型在不同子任务上的表现;
同时结合DICE系数衡量细胞核与背景的分离情况,用AJI与以往研究对比。
对于细胞核分类评估,定义 F c t F_{c}^{t} Fct分数综合考虑检测和分类任务,通过距离判断细胞核是否被正确检测。
1-4:实验结果
引入CoNSeP数据集,包含41个H&E染色的结直肠腺癌图像切片和24,319个注释细胞核。
在多个数据集上对比HoVer-Net与其他方法,结果显示HoVer-Net在细胞核实例分割上表现卓越,在多个数据集上PQ分数最高;在泛化能力研究中,HoVer-Net对未见数据有良好的适应性;
在分类实验中,相比其他方法,HoVer-Net在多个类别上取得了更高的 F 1 F_{1} F1分数,能有效实现同时实例分割和分类。
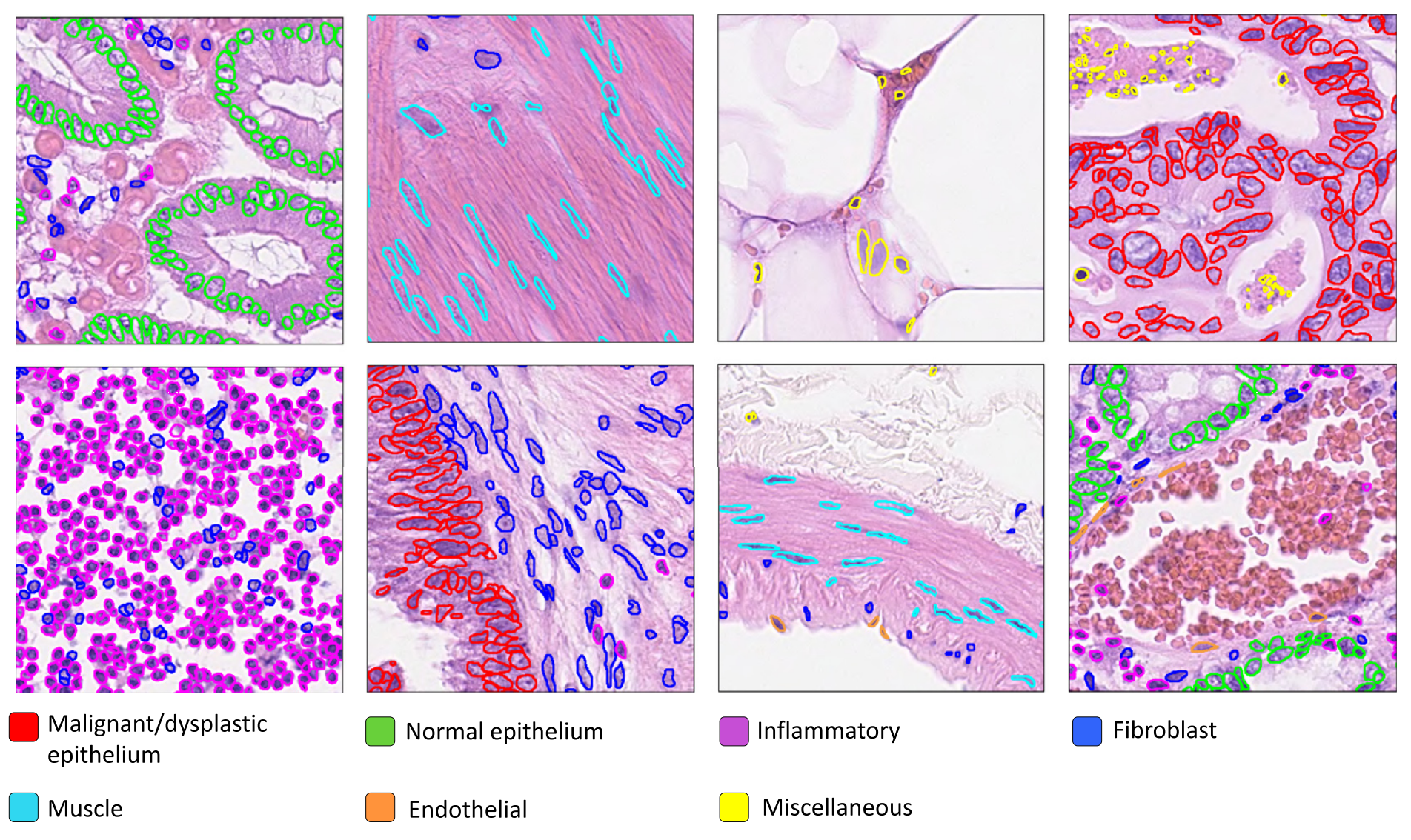
二、细胞核实例分割与分类
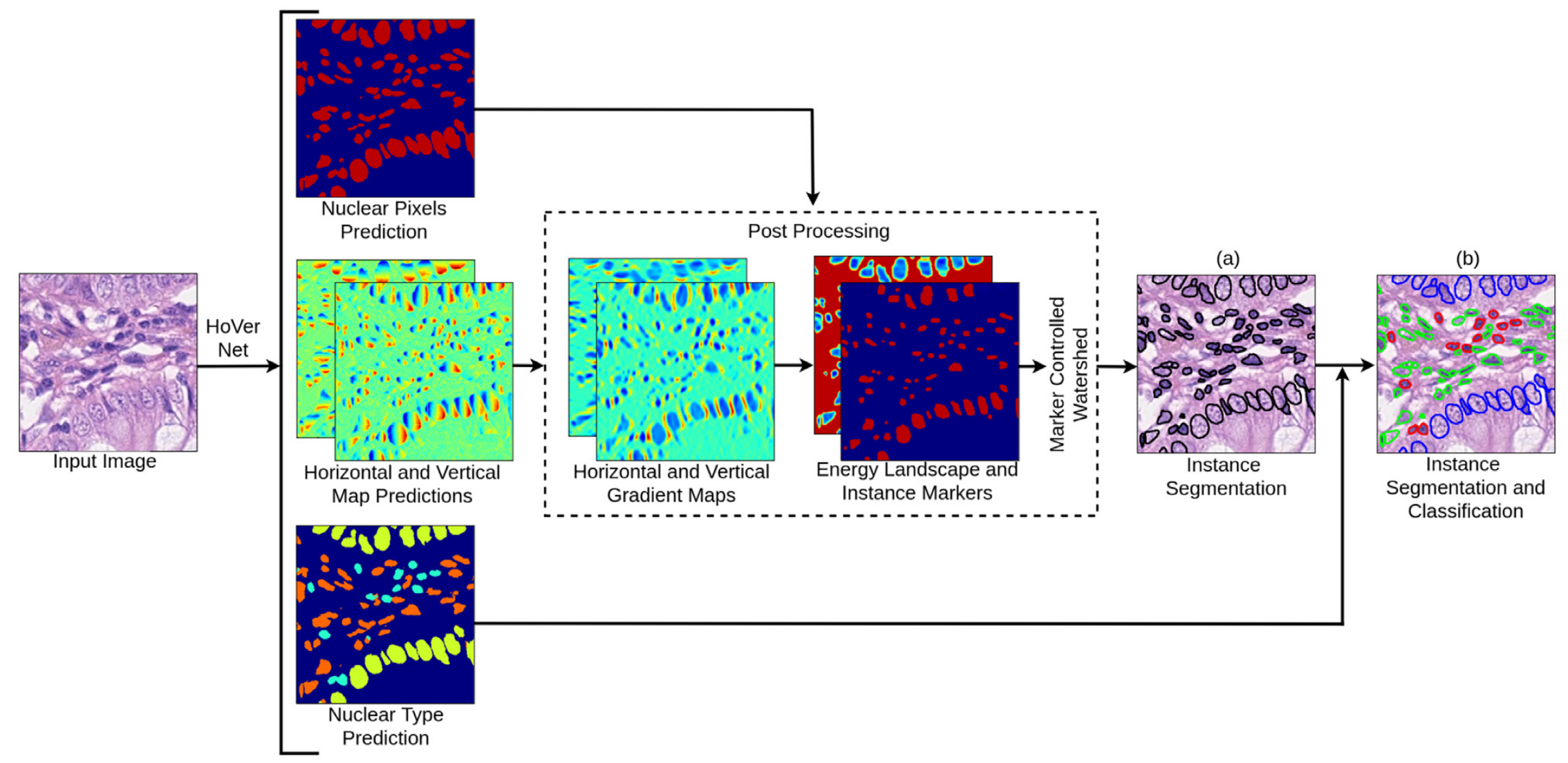
2-1:输入阶段
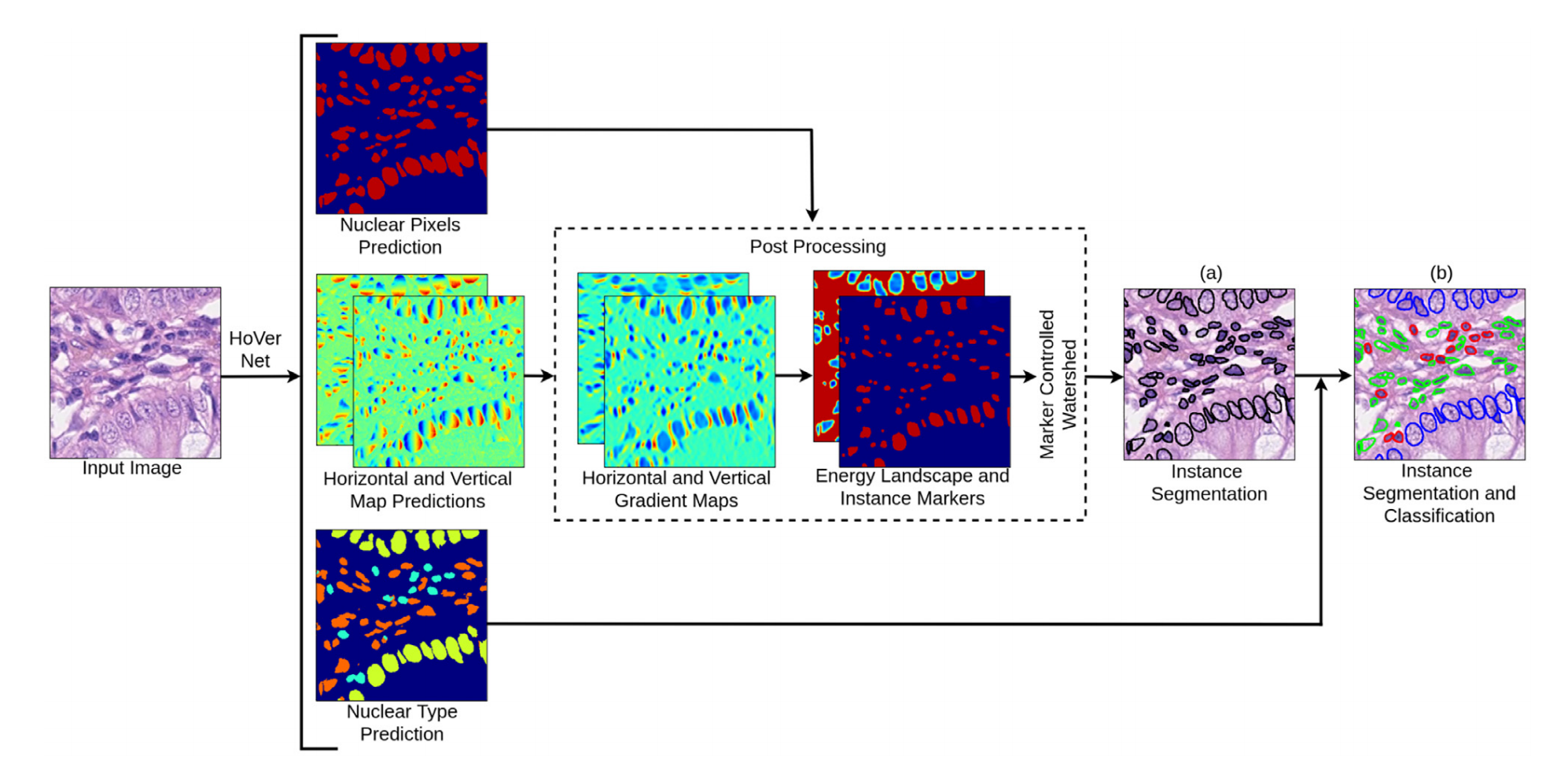
将组织学图像作为输入(Input Image),送入HoVer - Net网络。
这是整个流程的起始点,输入的图像包含了待分析的细胞核信息。
2-2:网络预测阶段
- 核像素预测(Nuclear Pixels Prediction) :网络输出核像素预测结果,判断图像中的像素是否属于细胞核,区分细胞核与背景 。
- 水平和垂直图预测(Horizontal and Vertical Map Predictions) :得到水平和垂直方向的图预测结果,计算核像素到质心的水平和垂直距离,为分离聚集的细胞核提供信息 。
- 核类型预测(Nuclear Type Prediction) :对细胞核类型进行预测,确定每个细胞核所属类别。
2-3:后处理阶段
- 梯度图生成(Horizontal and Vertical Gradient Maps) :基于水平和垂直图预测结果,计算水平和垂直梯度图,用于确定细胞核的边界信息 。
- 能量景观与实例标记(Energy Landscape and Instance Markers) :生成能量景观图并确定实例标记,结合核像素预测结果,为后续分割提供基础 。
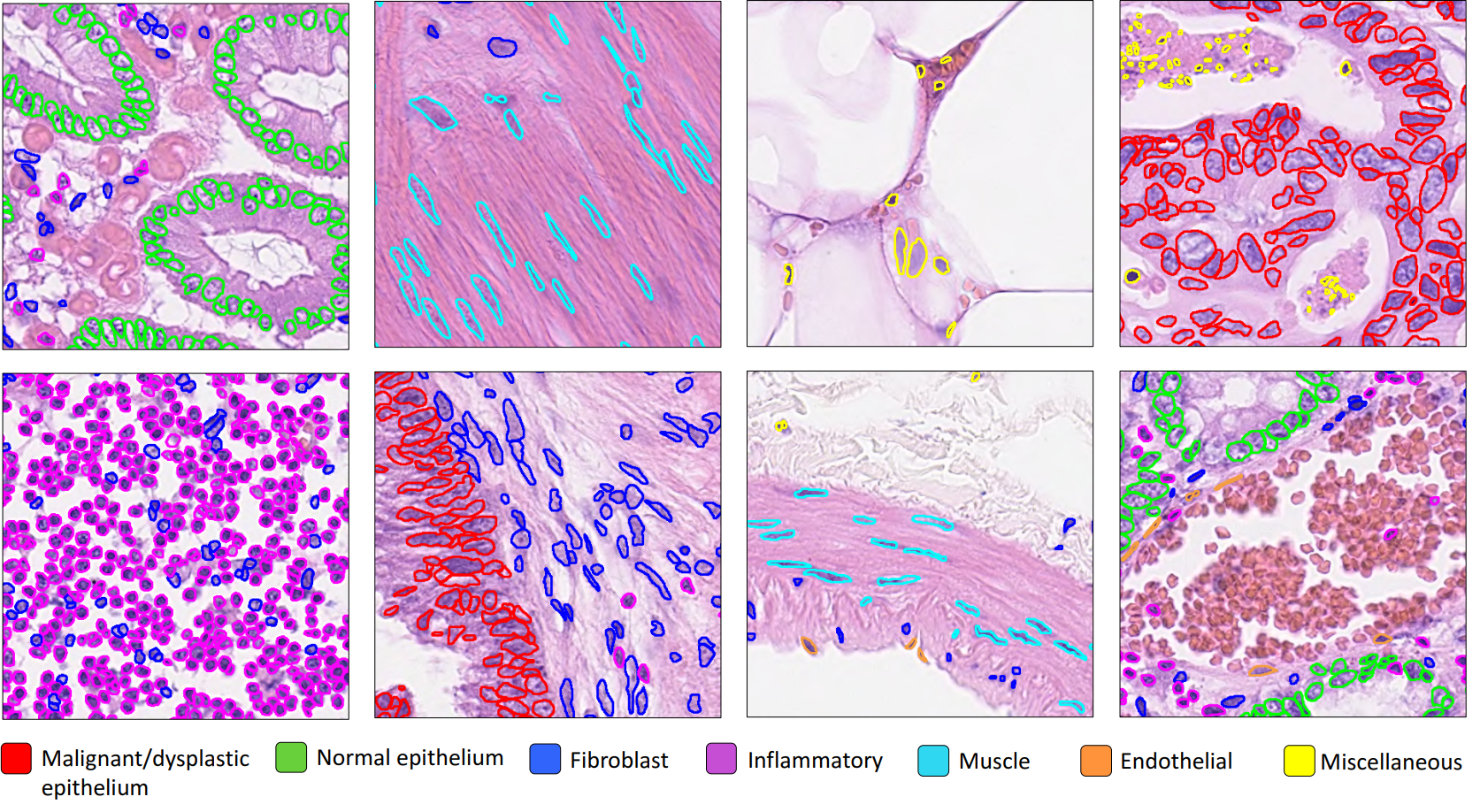
- 标记控制的分水岭算法(Marker Controlled Watershed) :通过该算法,利用前面得到的信息实现细胞核的实例分割(Instance Segmentation),得到单个细胞核的分割结果 。如果有细胞核分类标签,还能进一步实现实例分割与分类(Instance Segmentation and Classification),不同颜色的细胞核边界代表不同类型的细胞核 。
三、HoVer - Net网络架构图
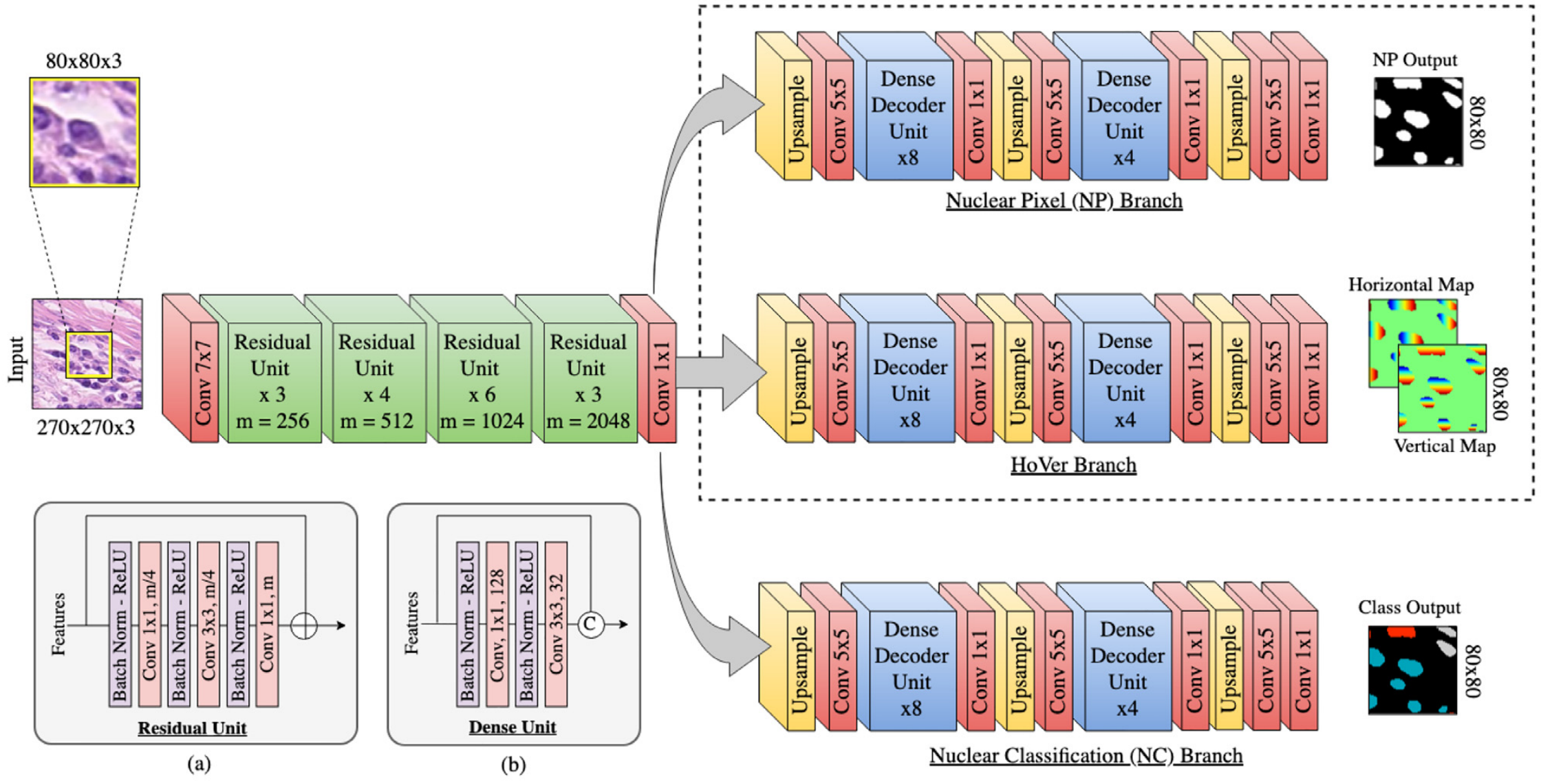
3-1:输入层
输入为尺寸 270 × 270 × 3 270×270×3 270×270×3的组织学图像(这里的 3 3 3代表RGB三个通道 )。
先通过一个 7 × 7 7×7 7×7卷积层,然后进入残差单元(Residual Unit)模块。
3-2:残差单元模块
包含多个不同参数设置的残差单元:
- 第一个残差单元模块有 3 3 3个单元,特征图数量 m = 256 m = 256 m=256 。
- 第二个残差单元模块有 4 4 4个单元, m = 512 m = 512 m=512 。
- 第三个残差单元模块有 6 6 6个单元, m = 1024 m = 1024 m=1024 。
- 第四个残差单元模块有 3 3 3个单元, m = 2048 m = 2048 m=2048 。
每个残差单元(图中(a)部分 )由批量归一化(Batch Norm)、修正线性单元(ReLU)和卷积层(Conv)组成,用于提取图像特征,且通过跳跃连接避免梯度消失问题,加深网络深度。
3-3:三个分支
- 核像素(NP)分支:用于预测核像素,判断图像中哪些像素属于细胞核。经过上采样(Upsample)、卷积(Conv)、密集解码单元(Dense Decoder Unit ,内部有多个重复结构 )等操作,最终输出尺寸为 08 × 08 08×08 08×08的核像素预测结果。
- HoVer分支:预测水平图(Horizontal Map)和垂直图(Vertical Map),计算核像素到质心的水平和垂直距离。结构与NP分支类似,也是通过上采样、卷积、密集解码单元等操作,得到相应的水平和垂直方向图,输出尺寸同样为 08 × 08 08×08 08×08 。
- 核分类(NC)分支:对细胞核类型进行分类。流程同样包含上采样、卷积、密集解码单元等操作,最终输出尺寸为 08 × 08 08×08 08×08的分类结果,不同颜色或标识代表不同细胞核类别。
3-4:密集单元
图中(b)部分展示了密集单元结构,也包含批量归一化、ReLU和卷积层,并且有特征拼接(C表示拼接操作 ),用于更高效地融合特征 。
整体网络通过这种结构,从输入图像开始,经特征提取后分三个分支分别完成核像素检测、距离预测和核类型分类任务,实现细胞核的同时分割与分类 。
四、环境配置
从这一部分开始,我们一起来复现Hovernet,这里使用的是作者团队后续开发的Tiatoolbox,可以直接调用Hovernet。
虽然已经把模型集成了,并且有详细的示例代码,但是配置环境以及应用到自己的数据上,仍然有一部分同学存在问题,所以这一期推送就带着大家把这些问题解决一下。
我们首先要做的就是,配置环境,这一部分Linux/Mac/Windows的都适用,尤其是Windows用户,这一期推送会很长一段时间伴随着你,因为你后续做其他的项目,配置环境遇到问题大概率会返回来看这篇推送!
4-1:克隆仓库
首先进入你想存放项目的地址,克隆仓库。
git clone https://github.com/TissueImageAnalytics/tiatoolbox.git
进入仓库
cd tiatoolbox
cd requirements
4-2:创建虚拟环境
Linux/Mac系统
PS:后续很多功能需要调用cuda,所以不建议使用Mac。
conda env create -f requirements.dev.conda.yml
Windows系统
由于windows系统比较特殊,导致我们很难一次性安装成功Pytorch和openslide,所以我们需要比其他系统的用户多完成一些步骤,否则你就会出错。
我们同样需要执行上面那行指令,不过我们执行之前需要对依赖项文件做一些变动,删除与Pytorch和openslide相关的选项。
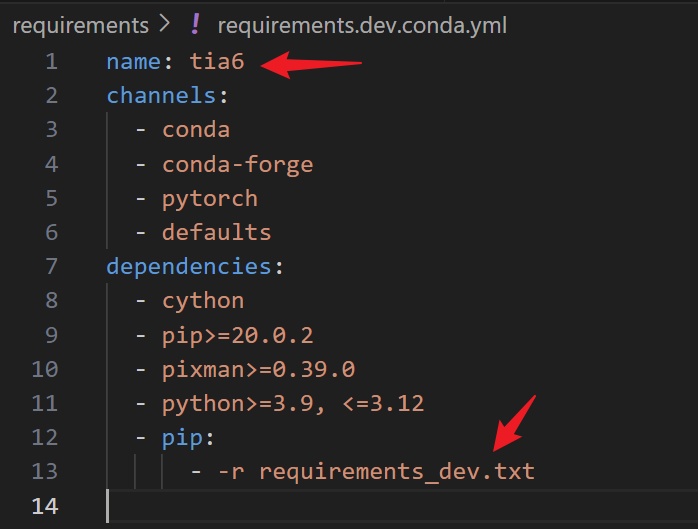
注意,依赖项文件基本都在requirements_dev.txt文件中,所以需要找到该文件修改。
完成了上述工作以后,我们就可以执行这行指令了。
conda env create -f requirements.dev.conda.yml
4-3:配置openslide
注意,这一部分是面向Windows用户的教程,其余系统的可以忽略。
1、下载和解压文件
首先进入官网下载界面,链接:https://openslide.org/download/
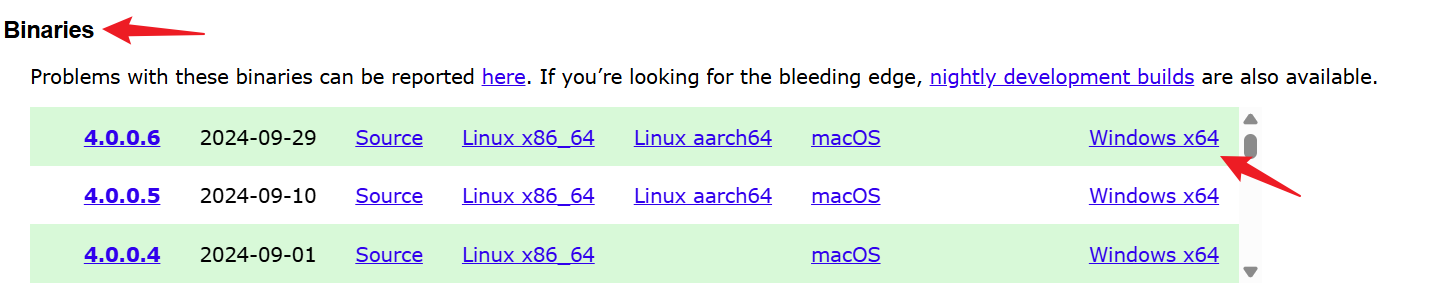
选择最新版本的二进制文件,下载并解压到anaconda的Library子目录里面。
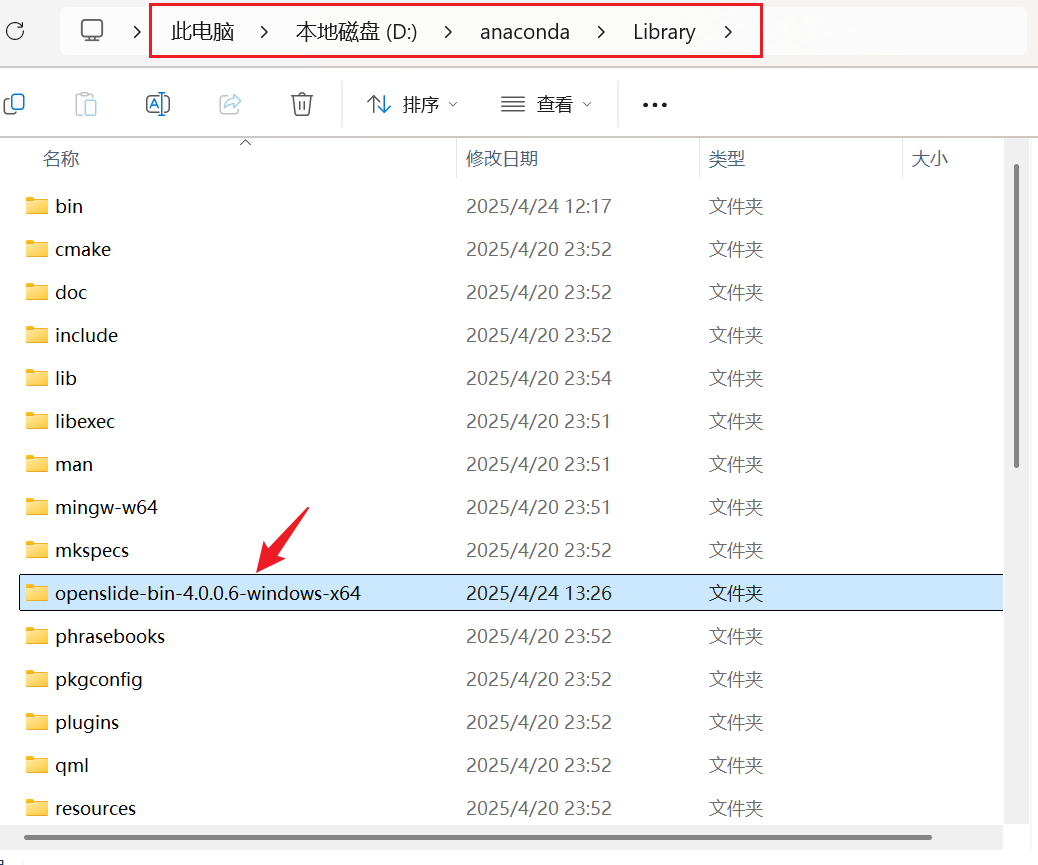
2、配置环境变量
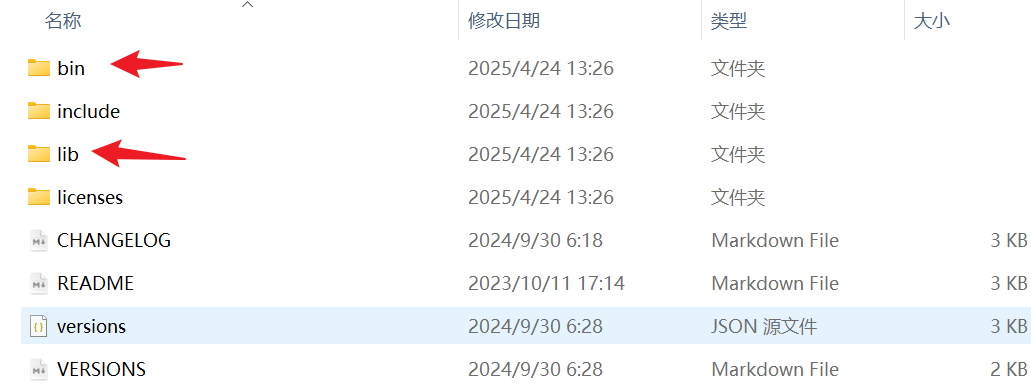
打开解压好的二进制文件,复制里面的bin和lib文件夹的文件路径。
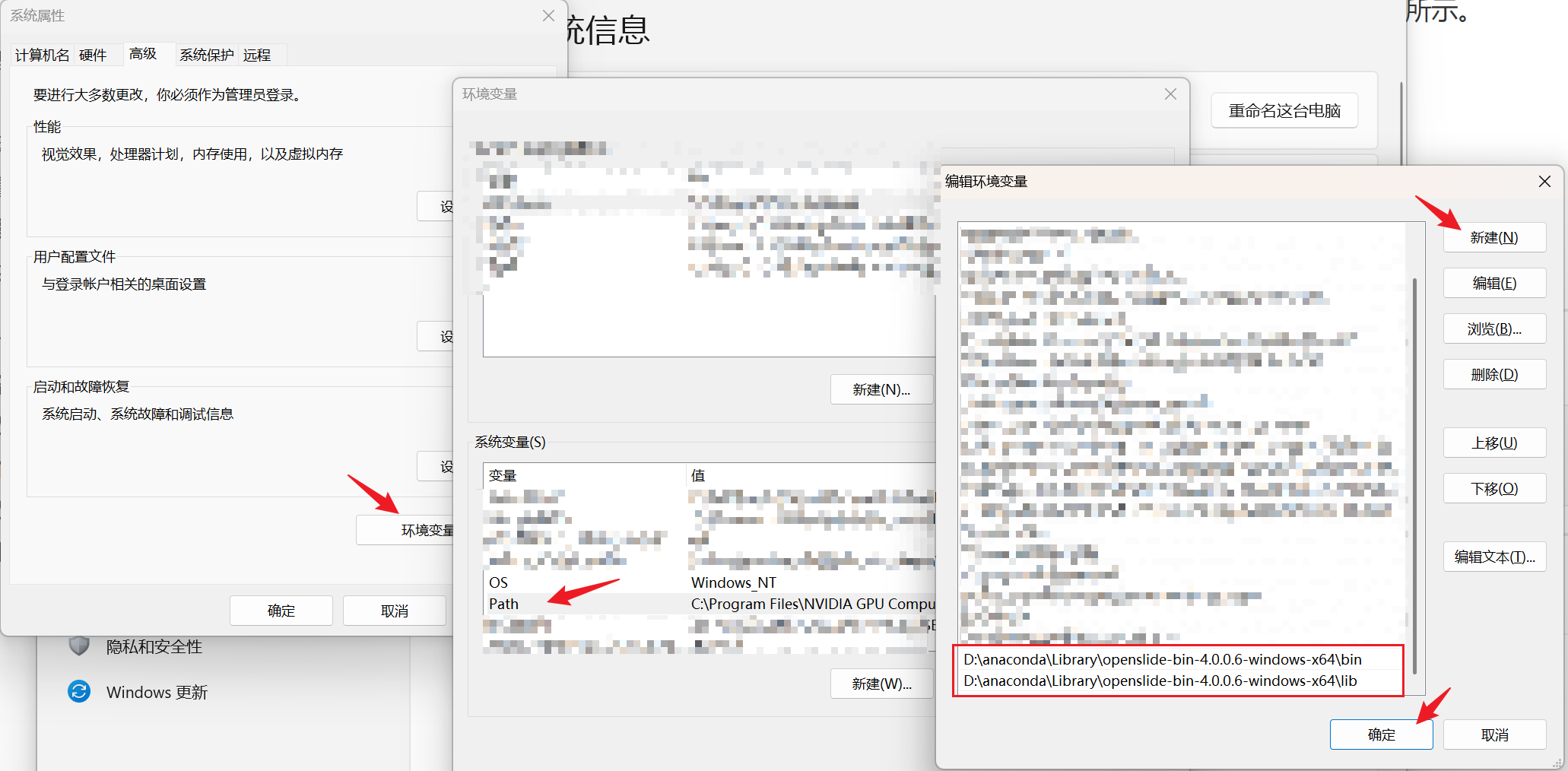
在系统变量中找到Path项,双击进入,编辑路径,将上面提到的两个路径粘贴至路径中,如图所示。
3、修改文件
进入我们刚刚建立的虚拟环境文件,找到lowlevel.py文件。
注意:更多内容请前往同名公众号/知识星球获取!
结束语
本期推文的内容就到这里啦,如果需要获取医学AI领域的最新发展动态,请关注小罗的推送!如需进一步深入研究,获取相关资料,欢迎加入我的知识星球!



















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









