







小罗碎碎念
在医学AI领域,数字病理图像中细胞核的精准分析对癌症诊断与预后极为关键。
这篇文章聚焦于此,提出了一种创新的深度学习框架。

该框架针对现有方法受限于数据集变异性、易陷入局部最优的问题,采用两级集成建模策略,同时对HoVer-Net架构进行改进,如更新编码器、优化解码器和实施模型正则化,旨在实现更准确且鲁棒的细胞核分析。
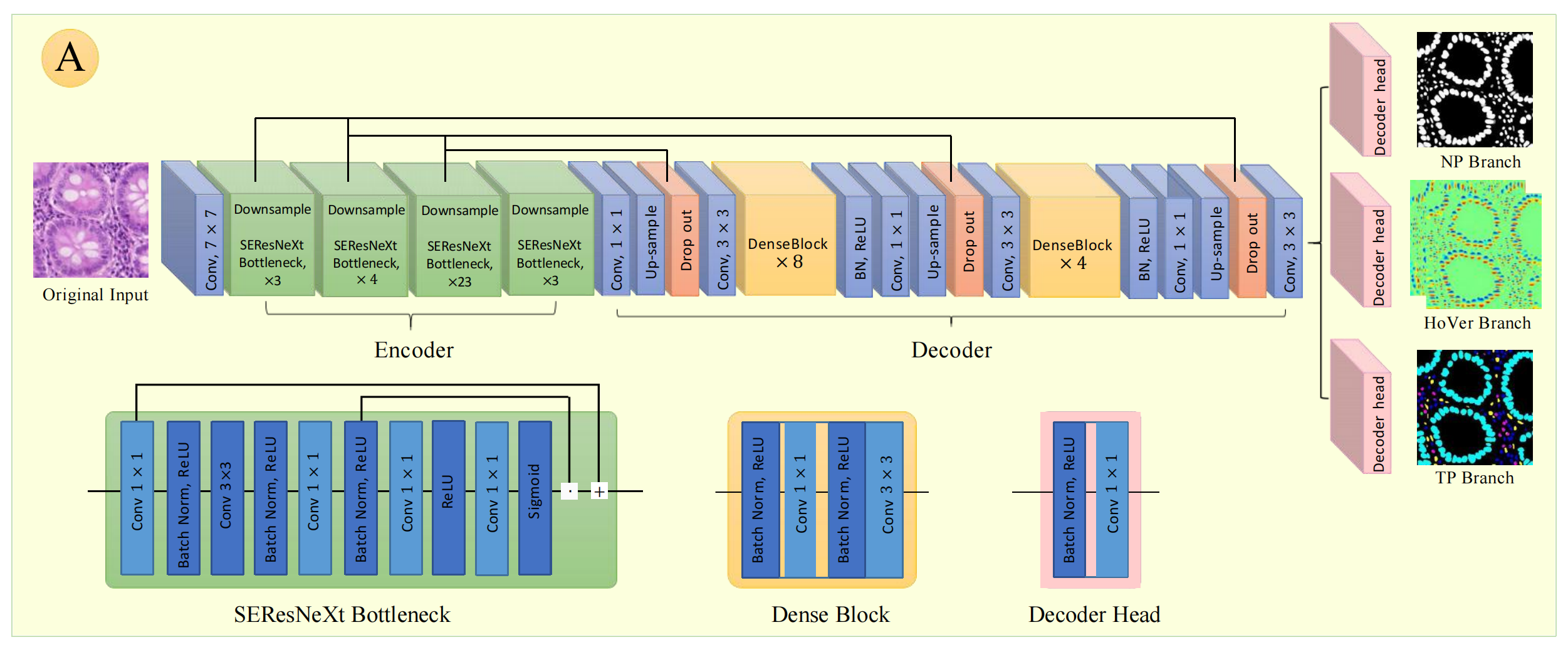
文中详细阐述了方法的各个关键部分。基础模型基于HoVer-Net构建,运用不同的编码器骨干,输出多种用于细胞核检测、定位和分类的特征图,并通过模型正则化和精心设计的复合多任务损失函数提升性能。
模型集成系统包含模型内集成和模型间集成,充分融合不同模型和输入增强带来的多样性优势。后处理阶段结合多种输出图完成细胞核实例的分割、分类及细胞组成预测。
实验部分使用多个多站点异质数据集训练和验证,采用 m P Q + mPQ+ mPQ+和 R 2 R^{2} R2等指标评估,结果显示该框架在多个公共基准测试中表现优异,在CoNIC 2022挑战赛中取得顶尖排名。
这篇文章为医学AI研究人员提供了极具价值的参考。其提出的框架和方法有效提升了细胞核分析的准确性和鲁棒性,在实际应用中可辅助病理诊断和治疗规划。
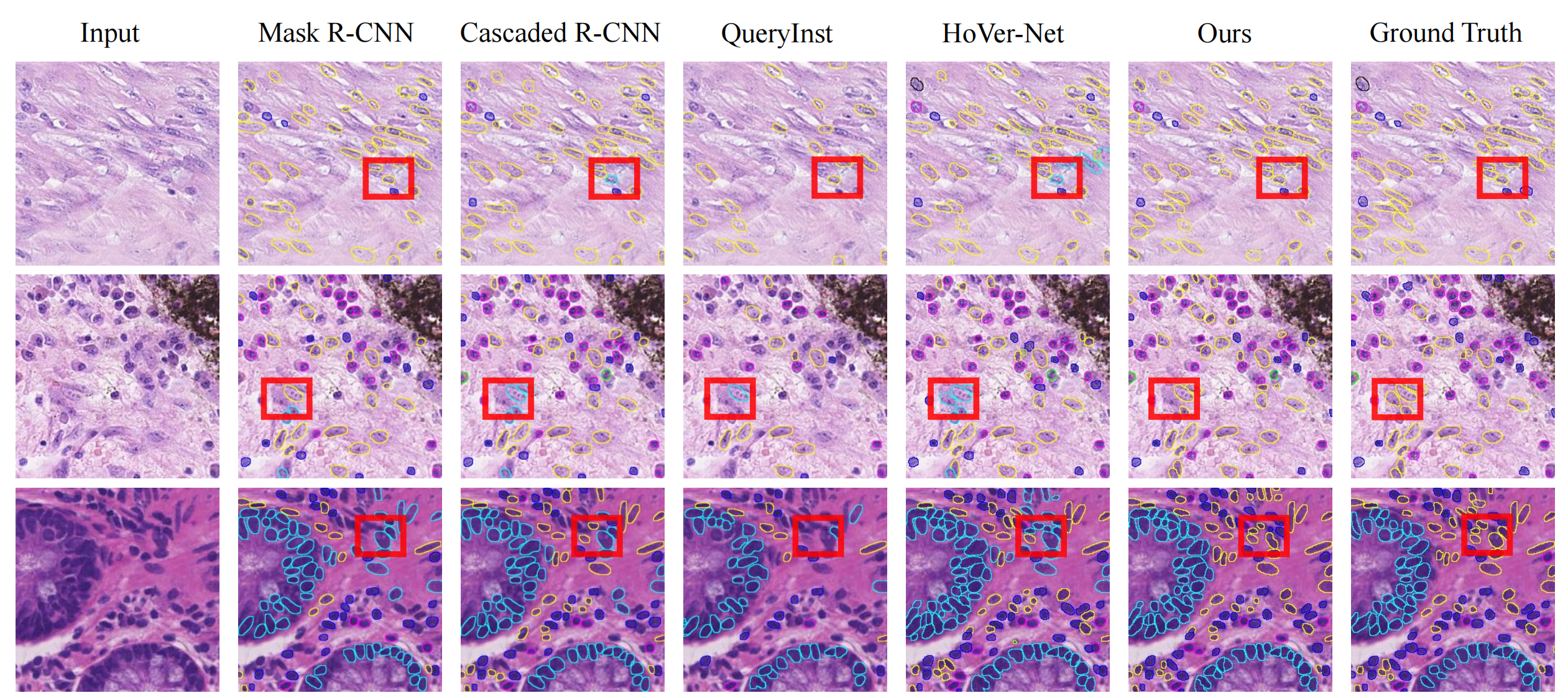
尽管存在计算复杂度较高的问题,但为后续研究指明了方向,研究人员可在此基础上进一步优化,探索如何平衡计算资源与模型性能,拓展该框架在更多医学影像分析任务中的应用,推动医学AI领域的发展。
交流群
欢迎大家加入【医学AI】交流群,本群设立的初衷是提供交流平台,方便大家后续课题合作。
目前小罗全平台关注量67,000+,交流群总成员1500+,大部分来自国内外顶尖院校/医院,期待您的加入!!
由于近期入群推销人员较多,已开启入群验证,扫码添加我的联系方式,备注姓名-单位-科室/专业,即可邀您入群。
知识星球
对推文中的内容感兴趣,想深入探讨?在处理项目时遇到了问题,无人商量?加入小罗的知识星球,寻找科研道路上的伙伴吧!
一、文献概述
“Keep It Accurate and Robust: An Enhanced Nuclei Analysis Framework”提出了一种用于数字病理图像中细胞核分割、分类和细胞组成预测的深度学习框架,通过集成建模和对HoVer-Net架构的改进,有效提升了模型性能,在多个公共基准测试中取得了优异成绩。

1-1:研究背景与目的
病理图像分析对癌症诊断和预后意义重大,其中细胞核分割、分类及细胞组成预测是关键环节。
现有方法受数据集变异性限制,易陷入局部最优。
本研究旨在提出新框架,通过集成建模提高模型鲁棒性,实现更精准的细胞核分析。
1-2:相关工作
回顾细胞核分割与分类、细胞组成预测及集成建模的相关研究。
现有细胞核分割与分类方法分为仅分割和同时分割分类两类,存在区域提议不准确、模型过拟合等问题;
细胞组成预测方法包括直接回归和先检测后计数两类;集成建模可结合多个模型优势,提高深度学习系统性能。
1-3:方法
- 基础模型:基于HoVer-Net框架构建基础模型,采用不同编码器架构,输出核像素(NP)图、水平/垂直距离(HoVer)图和核分类(NC)图。运用模型正则化策略,采用复合多任务损失函数优化模型。
- 模型集成系统:提出两级模型集成系统,包括模型内集成(对同一基础模型的不同增强输入进行预测并平均)和模型间集成(将不同基础模型的预测结果平均),以利用不同模型的优势。
- 后处理:利用HoVer和NP图进行分水岭分割,结合NC图通过像素投票对细胞核进行分类和计数,实现细胞核实例的分割、分类及细胞组成预测。
- 数据集:使用多站点异质数据集Lizard进行预训练,在PanNuke和MoNuSAC数据集上进行验证,以减少数据集偏差,训练更具泛化性的模型。
1-4:实验
- 评估指标:采用 m P Q + mPQ+ mPQ+评估细胞核实例识别和分类性能, R 2 R^{2} R2评估细胞核组成回归性能。
- 实验设置:使用SEResNeXt50和SEResNeXt101作为编码器骨干,在多GPU工作站上进行分布式并行训练,采用大量数据增强技术,通过网格搜索确定损失函数权重。
1-5:结果
- 与其他方法比较:在大多数数据集和评估指标上,该框架性能优于当前先进方法,在CoNIC 2022挑战赛中,细胞核组成预测排名第1,分割/分类排名第3。
- 消融研究:分析编码器骨干、数据增强策略、类别不平衡处理方法和模型集成技术等组件的重要性,确定了最佳模型架构和训练策略。
二、细胞核分析方法的整体流程
2-1:基础模型架构
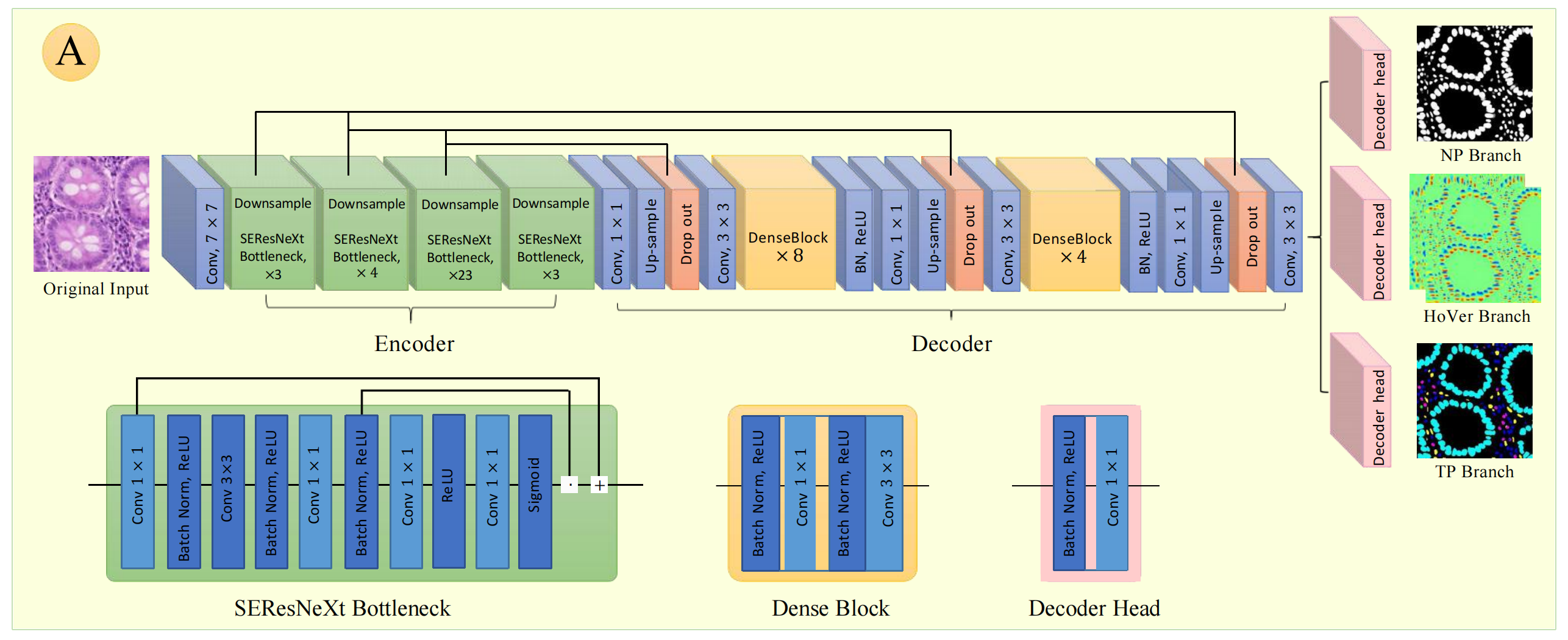
编码器(Encoder)
- 输入层:原始图像输入后,先经过一个 7 × 7 7×7 7×7卷积层,对图像进行初步特征提取。
- SEResNeXt Bottleneck模块:由多个SEResNeXt Bottleneck组成,分别有 3 3 3个、 4 4 4个、 23 23 23个、 3 3 3个,中间穿插下采样(Downsample)操作,逐步降低图像分辨率,增大感受野,提取更抽象的特征。SEResNeXt Bottleneck模块内包含 1 × 1 1×1 1×1卷积、批量归一化(Batch Norm)、ReLU激活函数、 3 × 3 3×3 3×3卷积等操作,通过残差连接(Residual Connection)提升网络训练稳定性和性能。
- 过渡层:经过一系列SEResNeXt Bottleneck模块后,通过 1 × 1 1×1 1×1卷积和上采样(Up - sample)操作,调整特征图维度,为解码器做准备。
解码器(Decoder)
- Dense Block模块:由多个Dense Block堆叠而成,图中有 8 8 8个一组和 4 4 4个一组。Dense Block内各层之间采用密集连接,能充分利用特征,提高特征复用率。模块中包含 3 × 3 3×3 3×3卷积、批量归一化(Batch Norm)、ReLU激活函数等操作。
- 上采样与输出层:经过Dense Block后,通过上采样(Up - sample)操作逐步恢复图像分辨率,再经过 1 × 1 1×1 1×1卷积和 3 × 3 3×3 3×3卷积调整通道数和特征图尺寸,最后通过Dropout防止过拟合,输出不同分支结果。
输出分支
- NP Branch:输出核存在(Nuclear Presence)相关特征图,用于判断图像中细胞核是否存在。
- HoVer Branch:输出水平/垂直距离(Horizontal - Vertical Distance )相关特征图,可用于确定细胞核位置和形状等信息。
- TP Branch:输出实例概率(Instance Probability)相关特征图,用于对细胞核实例进行概率预测。
2-2:模型内与模型间集成
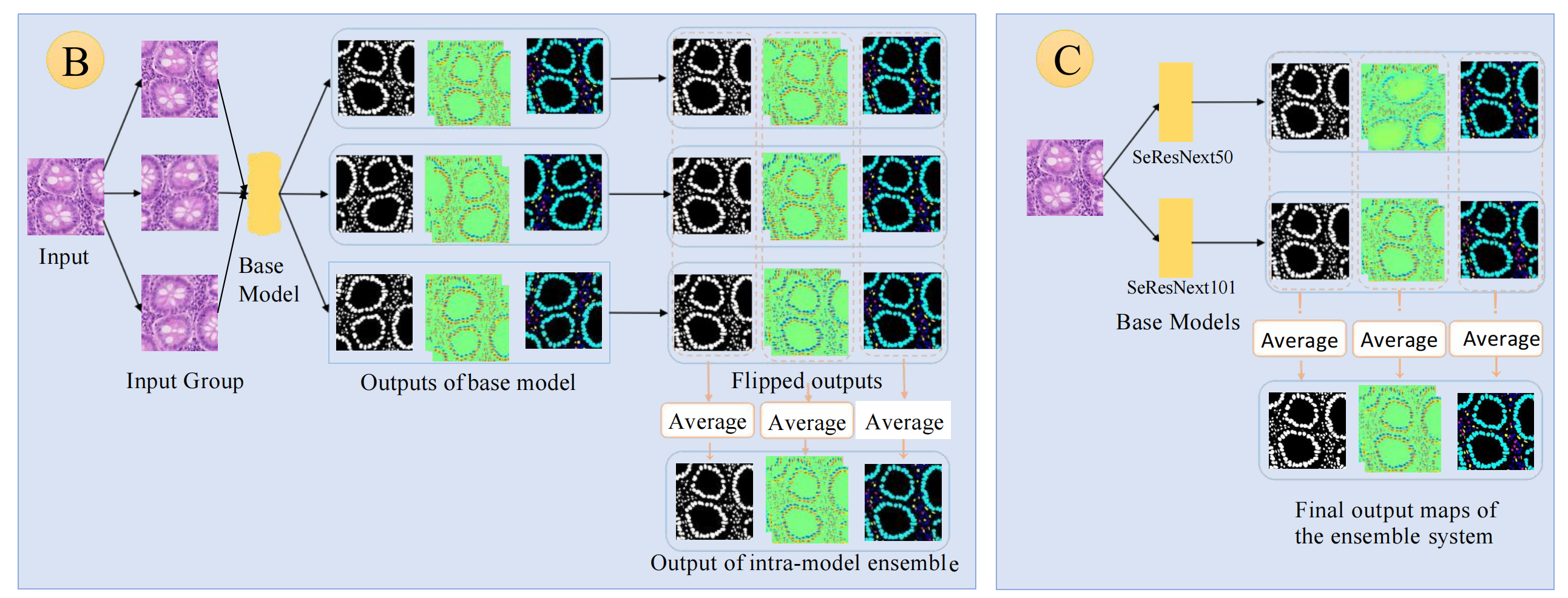
模型内集成(Intra - model ensemble)
- 原理:将原始输入图像进行一系列变换(如水平翻转、垂直翻转等),与原始图像共同组成输入组(Input Group)。这些图像输入基础模型(Base Model),得到相应输出(Outputs of base model)。之后将这些输出进行翻转操作得到翻转输出(Flipped outputs),再将原始输出和翻转输出对应位置的结果进行平均(Average),最终得到模型内集成的输出(Output of intra - model ensemble) 。
- 作用:通过这种方式,模型在推理时能利用输入图像的多个视角信息,增强对不同方向和形态细胞核的识别能力,提升模型鲁棒性,减少因图像方向等因素导致的误判。
模型间集成(Inter - model ensemble)
- 原理:使用不同编码器骨干(如这里的SeResNext50和SeResNext101 )构建多个基础模型(Base Models)。将同一输入图像分别输入这些基础模型,得到各自的输出。然后对这些不同模型的输出结果,在对应位置上进行平均(Average)操作,得到集成系统的最终输出特征图(Final output maps of the ensemble system) 。
- 作用:不同编码器骨干的模型具有不同的特征提取能力和侧重点,模型间集成可融合多个模型优势,弥补单个模型在特征提取上的局限性,进一步提高模型对细胞核分割、分类等任务的准确性和稳定性 。
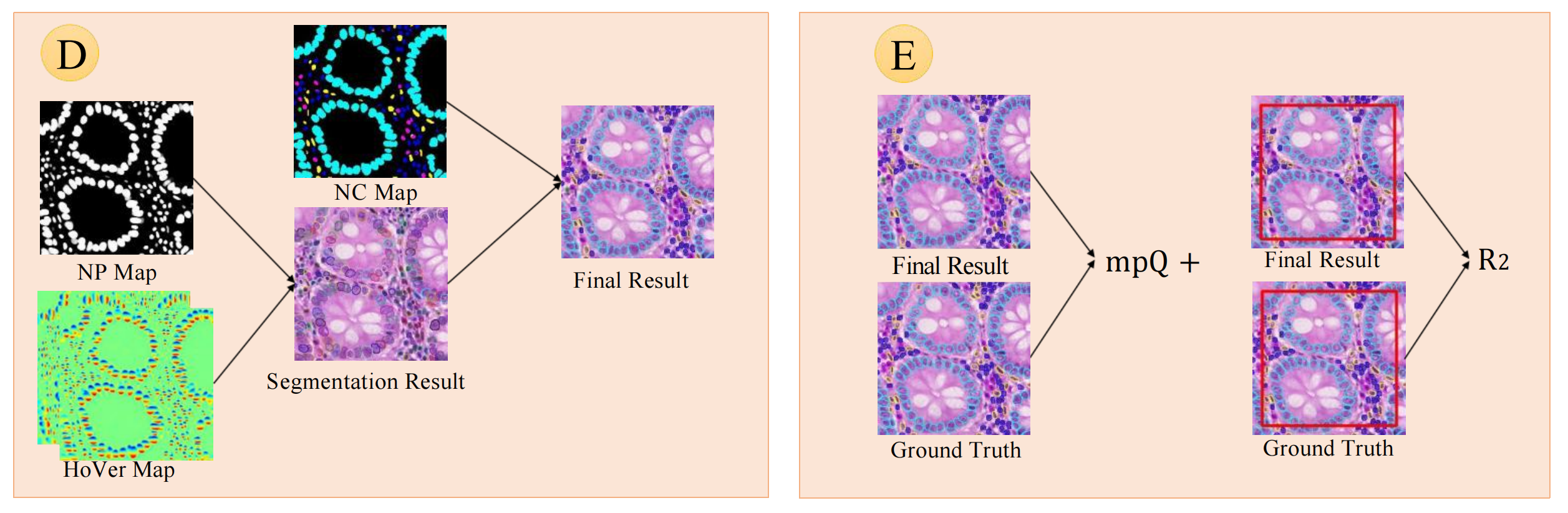
2-3:后处理与评估
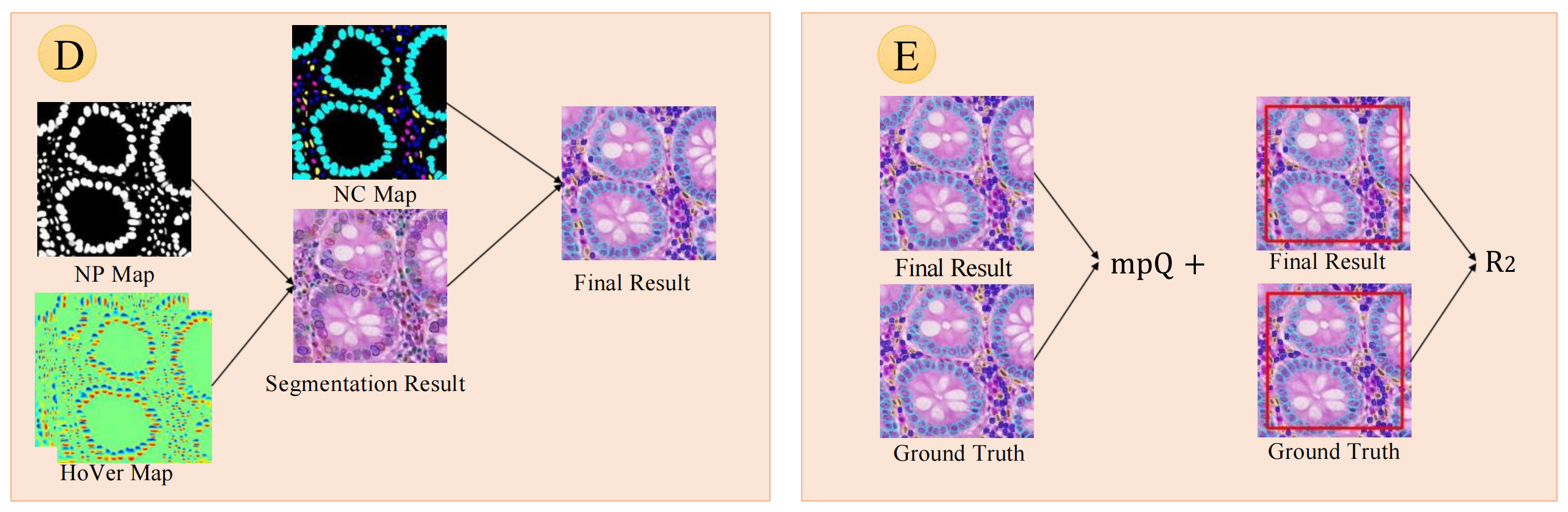
后处理流程
- 分割结果生成:利用核存在(NP)图和水平/垂直距离(HoVer)图,通过特定算法(如分水岭分割等)得到分割结果(Segmentation Result) ,确定细胞核在图像中的位置和轮廓。
- 最终结果整合:将分割结果与核分类(NC)图结合,通过像素投票等方式对分割出的细胞核进行分类,得到最终结果(Final Result) ,明确每个细胞核所属类别。
评估指标计算
- mPQ + 指标:将模型预测的最终结果(Final Result)与真实标注(Ground Truth)对比,计算多类全景质量(mPQ + )指标,衡量模型在细胞核实例识别和分类任务上的综合性能 ,该指标综合考虑了定位和分类的准确性。
- R 2 R^{2} R2指标:同样对比最终结果与真实标注,针对特定区域(图中用红色框标识)计算多类决定系数( R 2 R^{2} R2)指标 ,评估模型在细胞核组成回归任务中的表现,反映模型预测值与真实值的拟合程度。
三、项目复现概述
3-1:环境准备
依赖安装
# 基础环境
conda create -n nuclei_analysis python=3.8
conda activate nuclei_analysis
# PyTorch (根据CUDA版本选择)
pip install torch==1.12.1+cu113 torchvision==0.13.1+cu113 --extra-index-url https://download.pytorch.org/whl/cu113
# 项目依赖
pip install opencv-python scikit-image tqdm pandas
代码获取
git clone https://github.com/WinnieLaugh/CONIC_Pathology_AI.git
3-2:推理流程
-
预训练模型下载
-
下载包含以下内容的压缩包:
• 预训练权重 (checkpoints/)• 数据分割配置 (
splits/) -
解压至项目目录,保持文件结构一致:
├── checkpoints │ ├── model1.pth │ └── model2.pth ├── splits │ ├── train_split.csv │ └── val_split.csv -
执行推理
python scripts/eval_hv_ensemble_all.py \
--data_dir /path/to/test_data \
--model_dir checkpoints/ \
--output_dir results/
参数说明
• --data_dir: 测试数据路径(需包含images/和masks/子目录)
• --model_dir: 模型权重目录
• --output_dir: 输出目录(自动生成预测图和统计文件)
3-3:训练流程
-
数据准备
-
数据目录结构要求:
dataset/ ├── train │ ├── images │ │ ├── image1.png │ │ └── image2.png │ └── masks │ ├── mask1.png │ └── mask2.png ├── val │ └── ... (同train结构) └── test └── ... (同train结构) -
数据格式要求:
• 图像:PNG格式,建议尺寸256x256或512x512• 掩膜:单通道灰度图,像素值对应类别(0-背景,1-类别1,…)
3-4:分阶段训练
第一阶段:SE-ResNeXt50训练
python scripts/train_head_hv.py \
--encoder_name seresnext50 \
--pretrained_path pretrained/se_resnext50_32x4d-a260b3a4.pth \
--name hover_paper_pannuke_seresnext50_split_0 \
--split 0 \
--data_root /path/to/dataset \
--batch_size 16 \
--lr 1e-4
第二阶段:SE-ResNeXt101训练
python scripts/train_head_hv.py \
--encoder_name seresnext101 \
--pretrained_path pretrained/se_resnext101_32x4d-3b2fe3d8.pth \
--name hover_paper_pannuke_seresnext101_split_0 \
--split 0 \
--data_root /path/to/dataset \
--batch_size 8 \ # 减小batch_size防止显存不足
--lr 5e-5
关键参数说明
• --encoder_name: 骨干网络选择
• --pretrained_path: ImageNet预训练权重路径
• --split: 数据分割方案编号(对应不同交叉验证折)
• --num_classes: 类别数(默认自动检测)
结束语
本期推文的内容就到这里啦,如果需要获取医学AI领域的最新发展动态,请关注小罗的推送!如需进一步深入研究,获取相关资料,欢迎加入我的知识星球!



















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









