






metaGEM使用小记(解决各种问题)2024.2(三)
我的博客:小吴の博客
原文链接:metaGEM使用小记(解决各种问题)2024.1(三)
正式流程
这是原始文件夹省略了这些文件的内容,tree -I 'scripts|rules|envs'
每次新生成的文件和文件夹会用*标记出来
.
├── config.yaml
├── dataset
│ ├── L1EFG190305--AM43
│ │ ├── L1EFG190305--AM43_R1.fastq.gz
│ │ └── L1EFG190305--AM43_R2.fastq.gz
│ └── L1EFG190306--AM51
│ ├── L1EFG190306--AM51_R1.fastq.gz
│ └── L1EFG190306--AM51_R2.fastq.gz
├── logs
├── metaGEM.sh
├── Snakefile
├── temp
1.使用fastp质量过滤reads
每个样本提交一个质量过滤工作,每个过滤工作有2个CPU和20GB 内存,最大运行时间为2小时
bash metaGEM.sh -t fastp -j 2 -c 2 -m 20 -h 2 #运行主要程序(不要忘了source)
#可视化质量筛选结果:
bash metaGEM.sh -t qfilterVis
2.用 Megahit 组装
每个样品提交一个组装作业,每个组装作业有24个CPU和120GB 内存,最大运行时间为24小时:
bash metaGEM.sh -t megahit -j 2 -c 24 -m 120 -h 24
#可视化组装结果
bash metaGEM.sh -t assemblyVis
3. 使用 CONCOCT、 MaxBin2和 MetaBAT2分箱
使用 bwa 和 samtools,将每组成对的末端读数与每组组装的组合进行交叉映射,以获得样品间组合的丰度/覆盖率。每个样本提交一个作业,每个作业有24个CPU和120GB 内存,最大运行时间为24小时:
bash metaGEM.sh -t crossMapSeries -j 2 -c 24 -m 120 -h 24
- 注意这里如果报错找不到XXX.py是没有安装CONCOCT需要手动安装(好像后面还缺了什么文件先不管它)
wget https://github.com/BinPro/CONCOCT/archive/refs/tags/1.1.0.tar.gz
tar -zxvf 1.1.0.tar.gz
cd 1.1.0.tar.gz
python setup.py install
如果安装时 报错gsl/gsl_vector.h: No such file or directory
移步这里
或者快捷解决conda install anaconda::gsl
- 后面还让我装了pandas和pyarrow(奇怪)
conda install anaconda::pandas anaconda::pyarrow -y
注意: 旧的 rule CrosMap 被分成了 CrosMapSeries 和 rosMapParaller 两部分。运行序列映射更加简单,但是从计算资源的角度来看,对于有大量样本的数据集,例如 N = 1000,运行映射的代价可能会高得令人望而却步。
在样本之间运行每个binners,使用连续覆盖率:
bash metaGEM.sh -t concoct -j 2 -c 24 -m 80 -h 10
bash metaGEM.sh -t metabat -j 2 -c 24 -m 80 -h 10
bash metaGEM.sh -t maxbin -j 2 -c 24 -m 80 -h 10
4.使用metWRAP改进和重新组装
优化和重组bins
bash metaGEM.sh -t binRefine -j 2 -c 24 -m 150 -h 24
报错1(concoct):
TypeError: Feature names are only supported if all input features have string names, but your input has [‘int’, ‘str’] as feature name / column name types. If you want feature names to be stored and validated, you must convert them all to strings, by using X.columns = X.columns.astype(str) for example. Otherwise you can remove feature / column names from your input data, or convert them all to a non-string data type.
根据报错找到关键语句
concoct --coverage_file $(basename /home/gc/metaGEM/workflow/concoct/L1EFG190305--AM43/cov/coverage_table.tsv) --composition_file assembly_c10k.fa -b $(basename $(dirname /home/gc/metaGEM/workflow/concoct/L1EFG190305--AM43/L1EFG190305--AM43.concoct-bins)) -t 48 -c 800
找到了相关解决方案
找到/envs/metagem/lib/python3.10/site-packages/sklearn/utils/validation.py 文件
修改语句
feature_names = np.asarray(X.columns, dtype=object)
把这个语句注释掉然后添加
feature_names = np.asarray(X.columns.astype(str), dtype=object)
报错2(maxbin2):
/usr/bin/bash: line 28: run_MaxBin.pl: command not found
原因没有安装maxbin
下载 https://sourceforge.net/projects/maxbin/files/
wget https://sourceforge.net/projects/maxbin/files/MaxBin-2.2.7.tar.gz
tar -zxvf MaxBin-2.2.7.tar.gz
cd MaxBin-2.2.7/src
make
cd ..
apt-get install autoconf
./autobuild_auxiliary
在最后一步报错Cannot unzip bowtie2 zip file. Please make sure that [unzip] works properly.
原因:安装源下载网速过慢导致无法运行
修正方法:找到./MaxBin-2.2.7/buildapp文件,找到这一行
$cmd = "curl -L $URLBASE/$bowtie_f -k 1>$bowtie_f ";
注释掉并改成一下内容,注意后面有分号不要忘了
#$cmd = "curl -L $URLBASE/$bowtie_f -k 1>$bowtie_f ";
$cmd = "wget https://sourceforge.net/projects/bowtie-bio/files/bowtie2/2.2.3/bowtie2-2.2.3-source.zip"
不要忘了把新生成的文件夹加入环境变量
export PATH="/home/gc/MaxBin-2.2.7:$PATH"
在输入下列语句之前一定要先进入到metawrap 环境然后配置下面的功能
conda activate envs/metawrap
checkm_db="/home/ubuntu/checkm/"
echo ${checkm_db} | checkm data setRoot ${checkm_db}
如果不配置好会得到以下报错
It seems that the CheckM data folder has not been set yet or has been removed. Running: 'checkm data setRoot'.
Where should CheckM store it's data?
然后输入运行程序
bash metaGEM.sh -t binReassemble -j 2 -c 24 -m 150 -h 24
报错IOError: [Errno 2] No such file or directory: u’/home/gc/checkm/hmms/phylo.hmm’或者
It seems that the CheckM data folder has not been set yet or has been removed. Running: ‘checkm data setRoot’.
Where should CheckM store it’s data?
#先下载缺失的文件
cd ~
wget https://data.ace.uq.edu.au/public/CheckM_databases/checkm_data_2015_01_16.tar.gz
mkdir checkm
tar -zxvf checkm_data_2015_01_16.tar.gz -C ./checkm
checkm_db="/home/gc/checkm/"
echo ${checkm_db} | checkm data setRoot ${checkm_db}
终于在本地调试中发现了报错
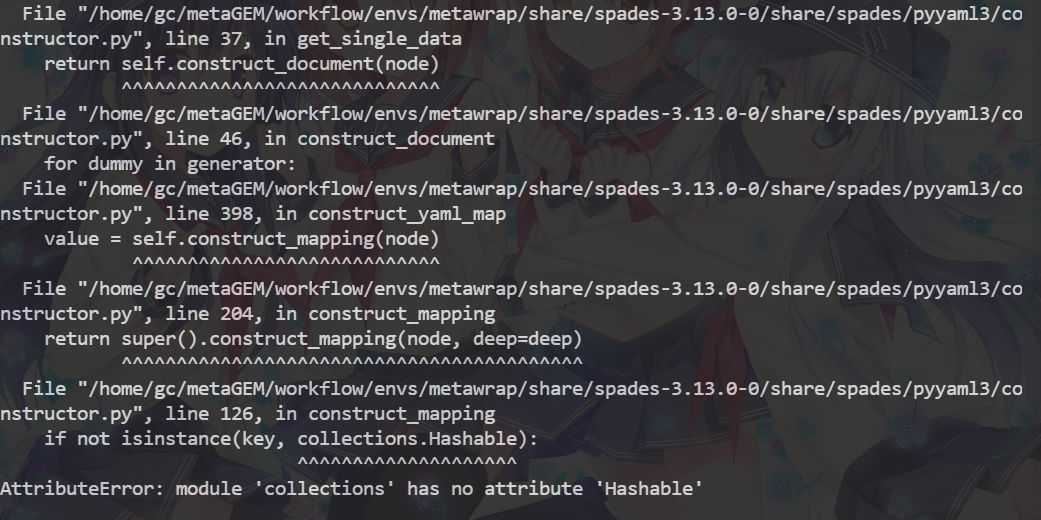
找到这个文件的这一行File "/home/gc/metaGEM/workflow/envs/metawrap/share/spades-3.13.0-0/share/spades/pyyaml3/constructor.py", line 126, in construct_mapping
找到代码 if not isinstance(key, collections.Hashable):
改成这样 if not isinstance(key, collections.abc.Hashable)
可视化输出
bash metaGEM.sh -t binningVis
报错*/*reassembled_bins/*.fa: No such file or directory
看reassembled_bins文件夹里面确实没有fa结尾的文件,这个嘶~,报错无从查起。那就再跑一遍bash metaGEM.sh -t binReassemble -j 2 -c 24 -m 150 -h 24这个命令吧。
可以看到在生成的中间文件中是有.fa结尾的文件的,而且还不少,因此我们监控一下在处理时发生了什么。
因为上次运行的时候有中断行为,因此重新运行一遍就好了
5用 GTDB-tk 进行分类
首先让我们从 metWRAP 重组输出中提取我们的 DNA bins:
bash metaGEM.sh -t extractDnaBins
运行 GTDB-tk 进行分类学分类:
bash metaGEM.sh -t gtdbtk -j 2 -c 24 -m 80 -h 12
在计算相对丰度之后,我们将可视化分类注释。
报错,没有数据库
conda uninstall gtdbtk
conda install -c conda-forge -c bioconda gtdbtk -y
download-db.sh
#检验是否安装成功
gtdbtk check_install
#如果报错The GTDB-Tk reference data does not exist or is corrupted
#则在Snakefile的1204行添加如下内容,并去掉注释
#安装的数据库在以下路径gc为你电脑的名字
/home/gc/metaGEM/workflow/envs/metagem/share/gtdbtk-1.7.0/db
export GTDBTK_DATA_PATH=/home/gc/metaGEM/workflow/envs/metagem/share/gtdbtk-1.7.0/db
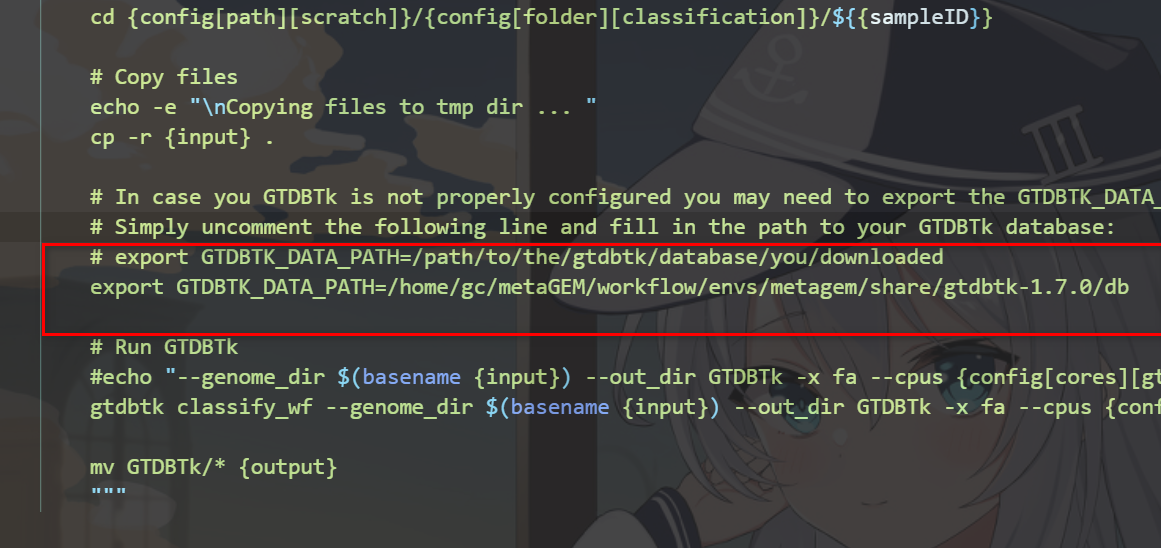
报错prodigal is not on the system path.
安装prodigal
wget https://github.com/hyattpd/Prodigal/releases/download/v2.6.3/prodigal.linux
wget https://github.com/hyattpd/Prodigal/archive/v2.6.3.tar.gz
tar -zxvf v2.6.3.tar.gz
cd Prodigal-2.6.3
sudo make install
报错hmmalign is not on the system path.
后面还有一些没有安装一块安装了
sudo apt install hmmer
conda install -c bioconda pplacer
#如果依赖报错则更改下列语句
#conda install python=3.6 #安装fastANI需要python3.6这里降一下版本
conda install -c conda-forge -c bioconda r-base=4 fastani
报错fastANI: error while loading shared libraries: libgsl.so.25
经过研究发现fastANI如果通过conda安装的话会报很多环境依赖的问题,因为在先前使用的是python3.10,网上有人说用python3.6才可以安装,于是我退回3.6发现依旧是有依赖问题无法安装,于是我寻求了手动编译安装的方法,如果有人
conda install -c bioconda fastani使用了上述语句成功安装,则不需要经过以下途径安装。
安装fastANI需要gsl依赖,在后面的安装中又出现了zlib库缺失的情况,我在这里就一块安装了
奇怪的是.so.25按道理来说应该用2.5版本的,但是2.5版本编译以后出来的是.so.23,下面有一个测试脚本,并无法运行,猜测应该是要用.so.25的文件,于是我又安装了gsl2.7,安装后出现了.so.25文件,并且编译没有出现问题(怪)
先安装gsl和zlib再安装fastani
#安装gsl
wget https://ftp.gnu.org/gnu/gsl/gsl-2.7.tar.gz
tar -zxvf gsl-2.7.tar.gz
cd gsl-2.7
./configure
make
sudo make install
vim /home/username/.bashrch
#在末尾添加
export LD_LIBRARY_PATH=$LD_LIBRARY_PATH:/usr/local/lib
export CFLAGS="-I/usr/local/include"
export LDFLAGS="-L/usr/local/lib"
#(验证)gsl是否安装成功使用后面的测试脚本
#安装zlib
wget https://www.zlib.net/zlib-1.3.1.tar.gz
tar -zxvf zlib-1.3.1.tar.gz
cd zlib-1.3.1
./configure
make
make check
sudo make install
#zlib的验证 zlib提供了测试样例,在examples里面
cd zlib-1.3.1/examples
gcc -c enough.c
gcc enough.o
./a.out #成功运行截图在后面
#安装fastani
wget https://github.com/ParBLiSS/FastANI/archive/master.zip
unzip master.zip
cd FastANI-master/
autoconf
./configure #--with-gsl=/usr/ 因为我们已经把gsl加入到环境变量里去了,所以这里我们把后面的语句注释掉
make
#验证fastani是否安装成功
fastANI
#返回Provide reference file (s)
测试脚本
//test.c
#include <stdio.h>
#include <gsl/gsl_sf_bessel.h>
int
main (void)
{
double x = 5.0;
double y = gsl_sf_bessel_J0 (x);
printf ("J0(%g) = %.18e/n", x, y);
return 0;
}
gcc -c test.c
gcc test.o -lgsl -lgslcblas -lm
./a.out

zlib的验证
6 bwa 和 samtools 计算相对丰度
bash metaGEM.sh -t abundance -j 2 -c 24 -m 80 -h 12
可视化分类和相对丰富度:
bash metaGEM.sh -t compositionVis
报错Error in library(tidytext) : there is no package called ‘tidytext’
发现这个tidytext是一个R包,安装R包环境
先安装R包环境
wget https://cran.r-project.org/src/base/R-4/R-4.3.2.tar.gz
tar -zxvf R-4.3.2.tar.gz
cd R-4.3.2
#! 编译,指定安装目录
./configure --prefix='/home/gc/R/R-4.2.2/' --enable-R-shlib=yes --with-readline=yes --with-libpng=yes --with-x=yes --with-blas --with-tcltk --with-pcrel
#! 安装
make&&make install
在安装其中的依赖
R#进入R环境然后输入下列语句
install.packages(c("mnormt", "psych", "SnowballC", "hunspell", "broom", "tokenizers", "janeaustenr"))
install.packages("tidytext")
以下是文件树
tree -I 'env|.snakemake|temp' > file-tree.txt














 798
798











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









