






本文是对《Algorithms for Detecting Significantly Mutated Pathways in Cancer》(2011年)的学习笔记
摘要:
近期基因组测序的研究表明导致癌症发展的体细胞突变分布在大量的基因中。这种变异的异质性使得努力从分散的乘客突变区分功能性突变变得复杂。假如癌症变异目标在细胞信号相对少且正常的路径上,则常规做法是估计是否已知路径涵盖了突变基因。我们介绍一种可供选择的方法在全基因组范围的基因相互作用网络中去检测突变基因。我们介绍一种计算高效的策略,该策略重新定义了在统计显著的患者中突变的交互网络的子网络。这个框架包含了两个主要成分。第一,我们使用在交互网络中的扩散过程去定义网络中每个突变基因的"局部影响邻居";第二,我们导出一个两阶段多假设检验约束错误发生率(FDR)来定义子网络。我们在庞大的人类蛋白质-蛋白质交互网络上使用来自胶质母细胞瘤和肺腺癌样本的体细胞突变数据测试这些算法。我们成功地恢复了这些癌症中已知的重要路径,并且定义了那些与其他癌症有牵连但不是以前报道的突变的额外路径。我们希望,我们会发现越来越多地使用使得癌症基因组研究的规模和范围不断增加。
一、数学模型(模型准备)
1.G = ( V,E )表示交互信息图
其中,顶点V表示个体患者(或相对应的基因),边E表示蛋白质-蛋白质关系或蛋白质-DNA关系。
2.Τ⊆V,表示Τ是在V中已经检测到的或是鉴定过的基因子集。(此处V表示患者对应的基因组集)
3.S为样本集。
4.g表示单个基因,对于每一个g,要么是突变的(mutated),要么是正常的(normal)。
5.Mi表示第 i 位患者在基因子集T中的突变基因子集,i = 1...|S|。
注:区分T与Mi,T为已知基因子集,Mi为在T中突变基因子集,Mi⊆Τ⊆V。
6.Sj是突变基因gj⊆T所归属的所有样本集(gj表示在基因子集T中的第j个基因),j = 1...|T|。
注:换句话说,Sj表示该样本集中的所有样本在基因子集T中第j个基因都发生突变。
7.m=Σi |Mi|,表示所有样本中可以观察到的变异基因总数。
8.路径(pathway)与子网络(subnetwork)是G的链接子图。
9.如果路径中任何一个基因是突变的,则都认为路径是突变的。
注:任何(any而不是every),指如果有一个或一个以上的突变,则认为路径是突变的
二、影响力图(Influence graph)
目的:识别样本中关于突变基因有效的子网络
有效性来源:(1).在网络基因中突变样本的个数 (2).在整个网络拓扑结构子网络基因的交互关系。
影响力(influence)计算:通过流动过程(diffusion process)测量结点s与其他所有结点的影响力。
1.流体以恒定速率被泵送如源结点s,通过图中的边进行流动。
2.流体以一个恒定的一阶速率γ从每一个结点流失。
3.
4.
5.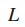
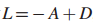
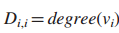
则,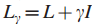
6.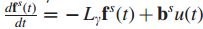
其中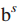
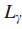
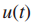
当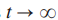
7.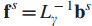
-------以上为准备过程,是前人已有的结论-------
-------以下为作者的影响力模型-----------------------
8.
9.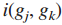
注意这并不一定是对称的,例如,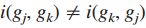
10.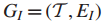
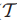
其中,边(gj,gk)为w(gj,gk) = min[ i(gk,gj) , i(gj,gk) ]。
11.n为交互网络结点的总数,则计算G1的花费主要来自于
三、结合模型发现有效子网络
发现有效子网络的方法是在给定的影响力图G1中识别结点集合,这就对图G1有两个要求
(1)通过高影响力的边缘连接;
(2)对应于一个有显着数量的样品中的突变基因
1.固定阈值δ,并得出一个移除权值w(gi,gj)<δ的边并且移除对应基因没有突变的结点的简化影响力图G1(δ)。
于是,发现有效子网络的问题被简化成了在G1(δ)识别连通子图。
在最大样本数中发现拥有k个突变基因的连通子图等价于下面的问题:连通最大覆盖问题(connected maximum coverage)
连通最大覆盖问题:
给定图G定义在n个顶点集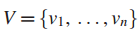
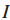
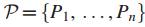

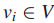
一个值k,在G中找到拥有k个结点的连通子图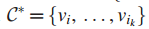
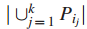
注: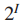
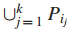
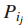
连通最大覆盖问题的实际解释:
接下来将上面的计算问题具体化。
用简化影响力图
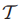
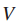
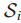
注:这里是样本种类最多,并不是样本个数最多。例如A={a b c d} B={a a a b b b b b},则A的样本种类为4,样本个数为4,B的样本种类为2,样本个数为8。
这个连通最大覆盖问题与最大覆盖问题是有关的。
最大覆盖问题:(也是一个NP-hard问题)
给定元素的集合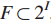
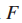
注意:这里的定义会与上面的定义有所分别,但大体解决的问题是一样的,只是连通最大覆盖问题把集合
由于以上两个问题说的是相同的事情,则连通最大覆盖问题也是一个NP-hard问题,就算是在简单的星状图中求解仍旧困难。
定理1 连通最大覆盖问题在星状图上是一个NP-hard问题。
因此,该问题转换为得到近似解。构建一个替代多项式时间算法,当最优解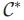
结合算法(替代多项式时间算法)如下:
为了获得一个解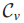
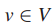
Step1 探测阶段
对每一个结点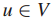
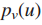
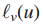
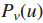

被
Step2 初始状态
算法从v结点建立连通图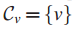

Step3 引入新结点
当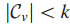
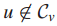
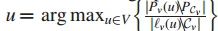
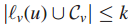
于是有新的解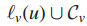
其中:
定理2 结合算法给予一个近似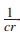
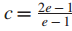
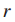
四、增强的影响力模型发现有效子网络
通过观察到的每一个突变基因的数量以及之后将相关增强影响力网络分解为连通分量,增强的影响力模型基于增强基因间的影响力测度。
Step1 定义增强的影响力图H;
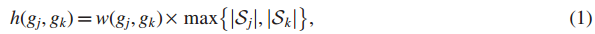
其中,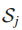
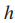
Step2 移除所有权值比阈值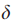
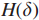
于是得到了图
五、统计分析
分析发现的连通图的有效性。
设计一个估计有效性的方法,通过使用在突变的基因分布上的任何一个空假设(null hypothesis)。
特别地,我们考虑在突变基因随机坐落在网络中的空假设分布(null hypothesis distributions),也就是当突变在网络拓扑中是独立发生的。
而其他发生在网络拓扑中非独立的突变分布也应该注意。
我们使用两个空假设分布:
1.第一个空假设分布
在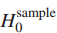
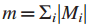
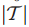
但无法解释在观察到的数据中,大量的突变都集中在少数基因上的情况。
2.第二个空假设分布
通过置换在网络中测试基因的分身实现。
选择一个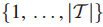
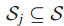
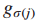
3.两阶段多假设检验(two-stage multi-hypothesis test)
一类错误:原假设是正确的,但却拒绝原假设。
二类错误:原假设是错误的,但却接受原假设。
令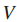
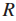
定义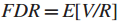
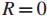
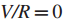
令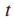
应用以上两张方法解决问题,发现的子网络被标记为统计有效的当且仅当p值是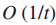
于是,发展一个两阶段多假设检验,用来标记数据中一些拥有较小的FDR值的统计有效的子网络。
令
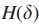
检测这些发现的有效性等价于同时检测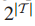
令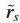
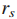
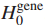
现在只检测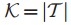
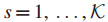
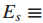
检测每一个置信水平在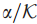
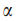
事实上规模大于s的连通子图个数统计上有意义并不意味着每一个连通子图有意义。因此加入第二个情况检测确保FDR上的上界。
定理3给定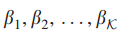
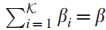
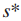
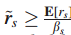
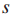
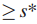
检测中,对于第i个最大的s,是使用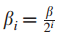
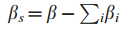
4.估计空假设的分布
增强影响模型:
假设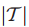
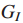
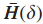
令M为观察到突变的基因个数,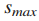
δ将图划分为一些连通子图,选择最大的δ,使任何图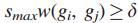
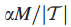
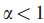
对于δ的这个选择,在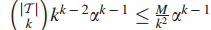















 8630
8630

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









