






周新洋
1
,
2
†
{ }^{1,2 \dagger}
1,2†,任永勇
2
†
{ }^{2 \dagger}
2†,赵倩倩
3
†
{ }^{3 \dagger}
3†,黄道一
1
{ }^{1}
1,王新波
1
,
2
{ }^{1,2}
1,2,赵婷婷
4
{ }^{4}
4,朱志兴
4
{ }^{4}
4,何文远
H
e
11
\mathrm{He}^{11}
He11,李淑媛
L
i
7
\mathrm{Li}^{7}
Li7,徐燕
X
u
7
\mathrm{Xu}^{7}
Xu7,孙瑜
9
,
10
{ }^{9,10}
9,10,余永国
Y
u
9
,
10
∗
\mathrm{Yu}^{9,10 *}
Yu9,10∗,吴盛楠
W
u
4
∗
\mathrm{Wu}^{4 *}
Wu4∗,王建
7
,
8
∗
{ }^{7,8 *}
7,8∗,余广军
Y
u
4
,
5
,
6
∗
\mathrm{Yu}^{4,5,6 *}
Yu4,5,6∗,何大可
H
e
11
∗
\mathrm{He}^{11 *}
He11∗,班博
3
∗
{ }^{3 *}
3∗,卢晖
L
u
1
,
2
,
4
∗
\mathrm{Lu}^{1,2,4 *}
Lu1,2,4∗
1
{ }^{1}
1 上海交通大学生命科学技术学院生物信息学与生物统计系,上海 200240,中国
2
{ }^{2}
2 上海交通大学-耶鲁大学生物统计与数据科学联合中心,国家转化医学中心数字医学技术中心,上海交通大学,上海 200240,中国
3
{ }^{3}
3 济宁医学院附属医院内分泌科,济宁医学院,山东济宁 272029,中国
4
{ }^{4}
4 上海交通大学医学院附属儿童医院,上海 200062,中国
5
{ }^{5}
5 香港中文大学医学院第二附属医院,深圳及深圳市龙岗区人民医院,深圳 518172,中国
6
{ }^{6}
6 上海儿科精准医学大数据工程研究中心生物医学信息中心,上海交通大学医学院附属儿童医院,上海 200062,中国
7
{ }^{7}
7 国际和平妇幼保健院,上海交通大学医学院,上海 200003,中国
8
{ }^{8}
8 上海交通大学医学院附属上海儿童医学中心医学遗传科与分子诊断实验室,上海 200127,中国
9
{ }^{9}
9 上海交通大学医学院附属新华医院临床遗传中心,上海 200092,中国
10
{ }^{10}
10 上海儿科研究院,上海 200092,中国
11
{ }^{11}
11 上海交通大学医学院附属新华医院小儿神经科,上海 200092,中国。
† \dagger † 这些作者对本工作贡献相同。
- 对应作者。邮箱: huilu@sjtu.edu.cn
摘要
准确诊断孟德尔疾病对于精准治疗和植入前遗传诊断(PGD)辅助至关重要。然而,现有方法往往未能达到临床标准,或依赖于大量数据来构建预训练的机器学习模型。为了解决这一问题,我们引入了一种创新的LLM驱动多智能体辩论系统(MD2GPS),该系统以自然语言解释诊断结果。它利用语言模型将数据驱动和知识驱动智能体的结果转化为自然语言,并促进这两个专业智能体之间的辩论。该系统已在四个独立数据集的1,185个样本上进行了测试,平均将TOP1准确率从 42.9 % 42.9 \% 42.9% 提高到 66 % 66 \% 66%。此外,在一个具有挑战性的72例队列中,MD2GPS在12名患者中识别出潜在致病基因,诊断时间减少了 90 % 90 \% 90%。此多智能体辩论系统的每个模块中的方法也可替换,便于其适应其他复杂疾病的诊断和研究。
关键词:孟德尔疾病诊断,变异优先级排序,LLM,多智能体辩论
引言
流行病学研究表明,如果将先天性异常视为遗传负担的一部分,全球大约
5.3
%
5.3 \%
5.3% 至
7.9
%
7.9 \%
7.9% 的新生儿患有严重的遗传疾病,主要为单基因突变引起的孟德尔疾病
1
−
3
{ }^{1-3}
1−3。在线人类孟德尔遗传数据库(OMIM)显示,目前的遗传研究已确定了约6,400种具有独特表型特征的孟德尔疾病,涉及约4,500个基因
4
{ }^{4}
4。尽管采用了诸如表型分析、成像和测序技术等复杂方法,孟德尔疾病的诊断率仍然较低,介于30%至
40
%
5
−
7
40 \%{ }^{5-7}
40%5−7。此外,使用传统方法如全基因组测序(WGS)和全外显子组测序(WES),孟德尔疾病的平均诊断时间约为两年
8
{ }^{8}
8。即使经过生物信息学软件和临床医生的严格筛选,每个人仍携带数百个可能致病或意义不确定的突变
9
{ }^{9}
9。此外,许多罕见遗传疾病表现出重叠的表型,使得诊断过程更加复杂并增加了误诊的可能性
10
,
11
{ }^{10,11}
10,11。准确诊断至关重要,因为它在指导体外受精(IVF)的植入前遗传诊断(PGD)中起着关键作用。这不仅有助于防止遗传疾病在家庭中的传播,还对减少这些疾病在人群中的总体患病率有所贡献
12
−
16
{ }^{12-16}
12−16。
在过去十年中,开发了各种计算工具,通过整合表型观察和基因组测序数据来增强遗传疾病的诊断。例如Exomiser
17
{ }^{17}
17、LIRICAL
18
{ }^{18}
18、Phen-Gen
19
{ }^{19}
19 和 diseaseGPS
20
{ }^{20}
20 等工具使用统计方法评估基因变异与表型表现之间的相关性,从而促进候选致病基因的优先排序。这些工具的有效性很大程度上取决于所使用统计数据的可用性和质量。同样,eXtasy
21
{ }^{21}
21、Xrare
22
{ }^{22}
22 和 AMELIE
23
{ }^{23}
23 等工具利用机器学习技术,需要大量数据进行监督训练或微调以优化性能。尽管这些方法根据变异数据优先排序候选基因,但缺乏自然语言解释意味着遗传疾病的最终诊断仍需医生和科学家通过广泛的文献回顾进行进一步验证
20
{ }^{20}
20。辩论是科学讨论中不可或缺的重要组成部分,是解决争议的关键机制。它促使参与者从不同角度探讨问题,这在医学诊断中尤为重要
24
{ }^{24}
24。在多学科治疗(MDT)实践中,辩论是有效治疗复杂医疗状况和得出循证结论的关键
25
,
26
{ }^{25,26}
25,26。然而,在诊断算法中应用辩论的最大挑战在于不同方法的语义理解和推理技术的局限性。
最近,大型语言模型(LLMs)在辩论过程中显示出相当大的潜力,尤其是在受到提示工程技术支持时 27 , 28 { }^{27,28} 27,28。LLMs可以通过利用自然语言中的广泛关联生成相关见解并总结证据。然而,它们也有局限性,不像生物信息学工具那样,LLMs无法提供详细的计算生物学注释。这强调了需要一种协同方法,将LLMs的分析能力与数据分析工具的技术专长结合起来,以充分利用它们在科学辩论中的综合优势 24 , 29 { }^{24,29} 24,29。
在这里,我们介绍了MD2GPS(Medical Doctor 2 GPS),这是一种由LLM驱动的多智能体辩论系统,旨在诊断孟德尔疾病。我们在四个独立数据集的1,185个样本上验证了MD2GPS。与当前最先进的方法相比,MD2GPS在所有模拟数据集中均取得了最高的TOP1分数。我们进一步在
另外一组具有挑战性的72个病例中测试了MD2GPS。MD2GPS的表现始终优于其他方法,有可能作为一种可解释的方法用于孟德尔疾病的诊断。
结果
MD2GPS的构建
如图1所示,MD2GPS由三个不同的智能体组成:数据智能体、知识智能体和辩论智能体。数据智能体处理以变异呼叫格式(VCF)和人类表型本体(HPO)术语提供的遗传变异,数据智能体注释变异的影响,过滤掉潜在的良性变异,并使用集成的生物信息学排名算法对剩余变异的致病潜力进行排名。它还用自然语言解释基因功能及其致病排名的依据。同时,知识智能体阐释基因及其生理功能,使用嵌入的GPT-4.0大型语言模型对基因致病性进行排名。然后,辩论智能体评估前两个智能体提供的证据的一致性和准确性,最终提供最终排名及其理由。
在构建MD2GPS之后,我们对MD2GPS与其他四种最先进的方法(diseaseGPS、Xrare、Exomiser和LIRICAL)在四个不同数据集上进行了面对面比较(表1)。我们考虑了四个评估指标,并在来自四个不同数据集的1185名遗传疾病患者上进行了评估(表1)。评估指标(TOP1、TOP3和TOP5)衡量TOP排名基因是正确致病基因的累计百分比。这套指标提供了诊断工具在更广泛的TOP候选范围内识别正确基因的能力的梯度测量。
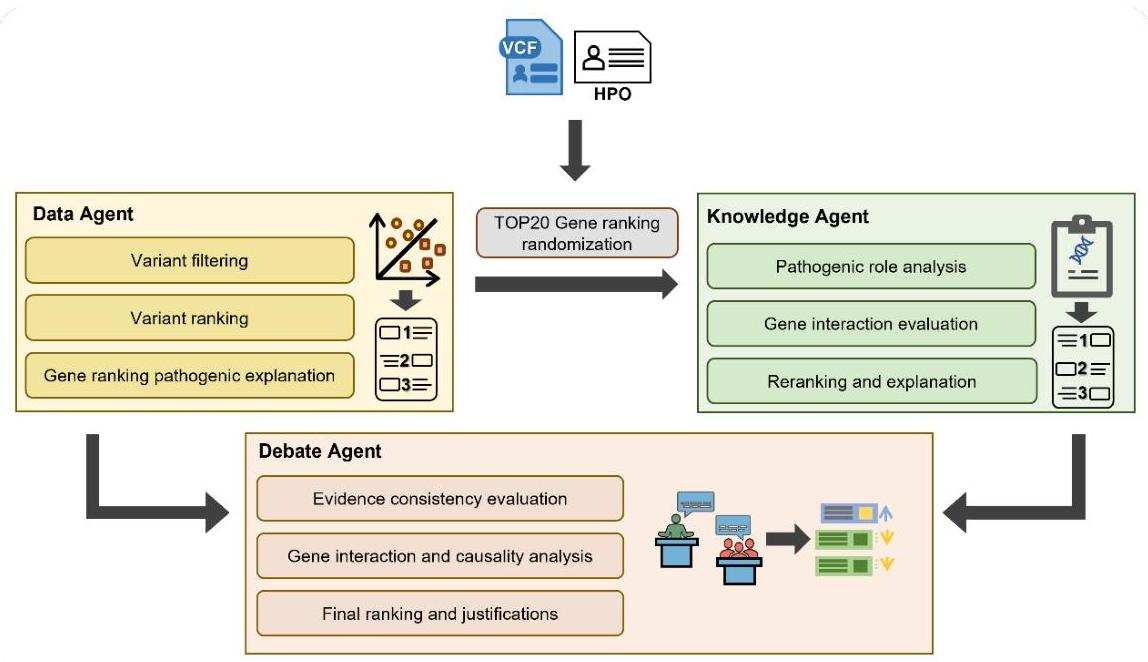
图1:MD2GPS的工作流程。MD2GPS集成了三个功能性智能体来处理遗传信息。数据智能体利用遗传数据,注释影响,过滤变异,并使用算法对致病潜力进行排名,同时提供自然语言解释基因功能。同时,知识智能体使用GPT-4.0模型进一步分析和基于生理功能对基因致病性进行排名。最后,辩论智能体评估其他智能体输出的一致性和准确性,最终提供最终的、有理由支持的基因排名。
MD2GPS与当前最先进方法的性能比较
图2总结了不同方法应用于诊断孟德尔遗传疾病时的性能。可以看出,MD2GPS在所有数据集中都获得了持续最高的TOP1得分,平均TOP1得分为 66.0 % 66.0 \% 66.0%,而其他方法得分较低,分别为 47.0 % 47.0 \% 47.0%(Xrare)、 42.9 % 42.9 \% 42.9%(diseaseGPS)、 34.0 % 34.0 \% 34.0%(LIRICAL)和 33.1 % 33.1 \% 33.1%(Exomiser)。此外,在TOP3和TOP5排名中,MD2GPS也表现出优越的性能。使用TOP5指标评估时,MD2GPS将TOP 5基因包括正确致病基因的累积百分比从 55.2 % − 73.2 % 55.2 \%-73.2 \% 55.2%−73.2% 提高到 85.1 % 85.1 \% 85.1%。
MD2GPS在所有数据集中始终优于其他方法,包括涉及单一或多重表型的模拟和真实案例情景中,均实现了最高的TOP1得分。在专注于矮小症单一表型的JN数据集中,MD2GPS实现了 70.0 % 70.0 \% 70.0% 的TOP1得分,超出表现次佳的方法Xrare 13.3 % 13.3 \% 13.3%(图2A)。在对734名发育障碍患者的分析中,MD2GPS再次实现了最高的TOP1得分,超越了表现次佳的方法diseaseGPS 8.2 % 8.2 \% 8.2%(图2B)。在涉及多种异常表型的情景中,MD2GPS进一步展示了其优越性。在一个以明确病因、诊断和治疗计划为特征的模拟数据集(RD数据集)中,它将TOP1准确率从 65.6 % 65.6 \% 65.6%(由LIRICAL实现)提高到 76.8 % 76.8 \% 76.8%(图2C)。在真实的SCH数据集中,一个多专家咨询数据集,MD2GPS将TOP1准确率从 39.6 % 39.6 \% 39.6% 提高到 65.8 % 65.8 \% 65.8%,增加了 26.2 % 26.2 \% 26.2%(图2D)。
我们进一步研究了MD2GPS和其他方法在RD数据集中按已知发病率分类的疾病诊断性能。根据中位发病率,这些疾病被分为四个不同的四分位数: < 1 / 125000 , [ 1 / 125000 , 1 / 75000 ] <1 / 125000,[1 / 125000,1 / 75000] <1/125000,[1/125000,1/75000], [ 1 / 75000 , 1 / 15000 ] [1 / 75000,1 / 15000] [1/75000,1/15000] 和 > 1 / 15000 >1 / 15000 >1/15000,MD2GPS在所有组中始终保持最佳的TOP1准确率。如图2E所示,在最低流行率四分位数( < 1 / 125000 <1 / 125000 <1/125000)中,MD2GPS达到了显著的 61.5 % 61.5 \% 61.5% TOP1准确率,明显优于其他方法。其表现高峰出现在下一个四分位数 [1/125000, 1/75000] 中,TOP1准确率达到令人印象深刻的 82.9 % 82.9 \% 82.9%,明显高于其他方法。对于中等流行率组 [1/75000, 1/15000] 的疾病,MD2GPS的TOP1准确率为 75.0 % 75.0 \% 75.0%,在此类别中表现最佳。在最高流行率类别( > 1 / 15000 >1 / 15000 >1/15000)中,MD2GPS继续领先,准确率达到 73.3 % 73.3 \% 73.3%,展示了其在不同遗传疾病流行率下的有效性。
表1. 孟德尔遗传疾病数据集。
| 数据集名称 | 来源 | 数据集描述 | 病例数 |
|---|---|---|---|
| DDD数据集 | 解码发育障碍(DDD)项目 | 我们去重了致病基因,并在DDD项目中每基因随机选择一个病例进行案例模拟。 | 734 |
| 发育障碍(DDD)项目 | |||
| RD数据集 | 手动构建自PubMed | 该数据集涵盖了“中国第一批罕见病名单”中列出的96种遗传疾病。 | 234 |
| SCH数据集 | 上海儿童医院 | 所有病例涉及复杂的多学科治疗场景,难以从现实世界中诊断。 | 187 |
| JN数据集 | 济宁医学院附属医院 | 一个真实的单一疾病矮小症数据集。 | 30 |
SCH和RD数据集涉及多种遗传疾病;SCH包括复杂的多学科治疗场景,RD涵盖“中国第一批罕见病名单”中列出的95种遗传疾病。JN和DDD数据集侧重于特定疾病表现:JN包含矮小症病例,DDD包括从解码发育障碍病例中去重的独特致病基因。
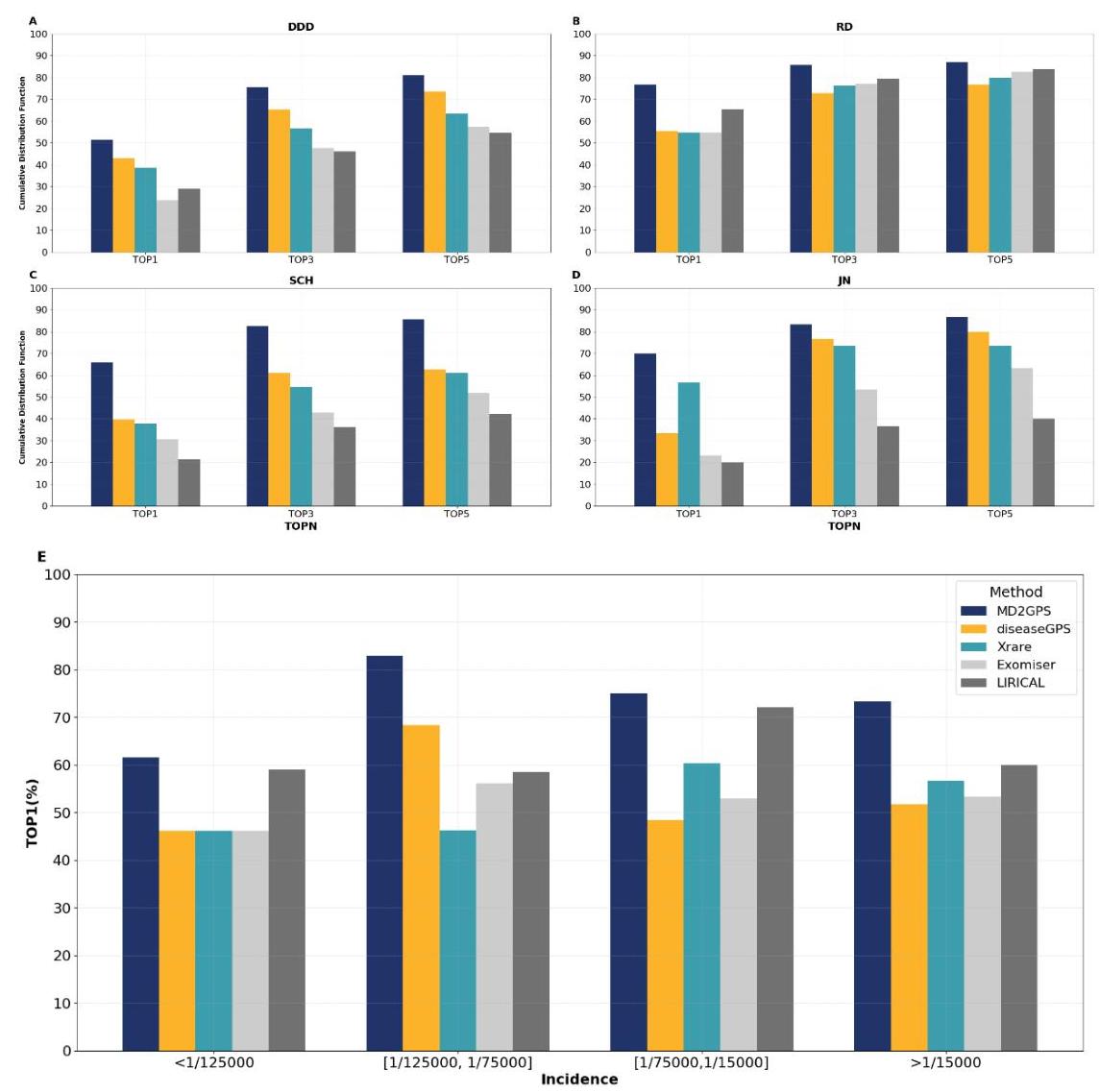
图2:四种孟德尔遗传疾病数据集应用诊断方法的性能。所有方法的性能评估通过两个模拟数据集A) DDD和B) RD以及两个真实数据集C) SCH和D) JN的累积分布函数(CDF)进行。E) 图表总结了MD2GPS与其他诊断工具在RD数据集上的比较性能,展示了不同发病率类别的TOP1准确率: < 1 / 25000 , [ 1 / 25000 , 1 / 75000 ] , [ 1 / 75000 , 1 / 15000 ] <1 / 25000,[1 / 25000,1 / 75000],[1 / 75000,1 / 15000] <1/25000,[1/25000,1/75000],[1/75000,1/15000], 和 > 1 / 15000 >1 / 15000 >1/15000。
评估辩论智能体在提高诊断准确性中的作用
为了评估辩论智能体对模型性能的影响,我们对知识智能体和辩论智能体的输出进行了比较分析。两者都利用先进的GPT-4大型语言模型对基因致病性进行排名。分析表明,辩论后诊断性能显著提高,TOP1准确率
相比知识智能体的平均值提高了
13.4
%
13.4 \%
13.4%。这种改进在现实生活中的复杂病例中尤为显著,正如SCH数据集所示,准确率提高了
16.6
%
16.6 \%
16.6%。这些发现突显了辩论智能体在通过更有效地评估证据的一致性和相关性来提高诊断准确性方面的重要作用。
我们将单一LLM驱动的知识智能体的诊断结果与多智能体辩论后获得的结果进行了比较。数据智能体的加入带来了必要的多样化视角,促进了深入辩论,而辩论智能体的引入则促进了战略提升。在多个数据集上的广泛测试表明,多智能体辩论显著增强了LLMs对遗传疾病的诊断能力(图3)。值得注意的是,最关键的预测(TOP1)的模型准确率提高了
10
%
10 \%
10% 到
16.6
%
16.6 \%
16.6%,而TOP5预测的稳定性得以保持。这些结果证实了多智能体辩论策略的有效性,并突出其对提高模型在识别关键诊断结果方面的精确性的重要贡献。
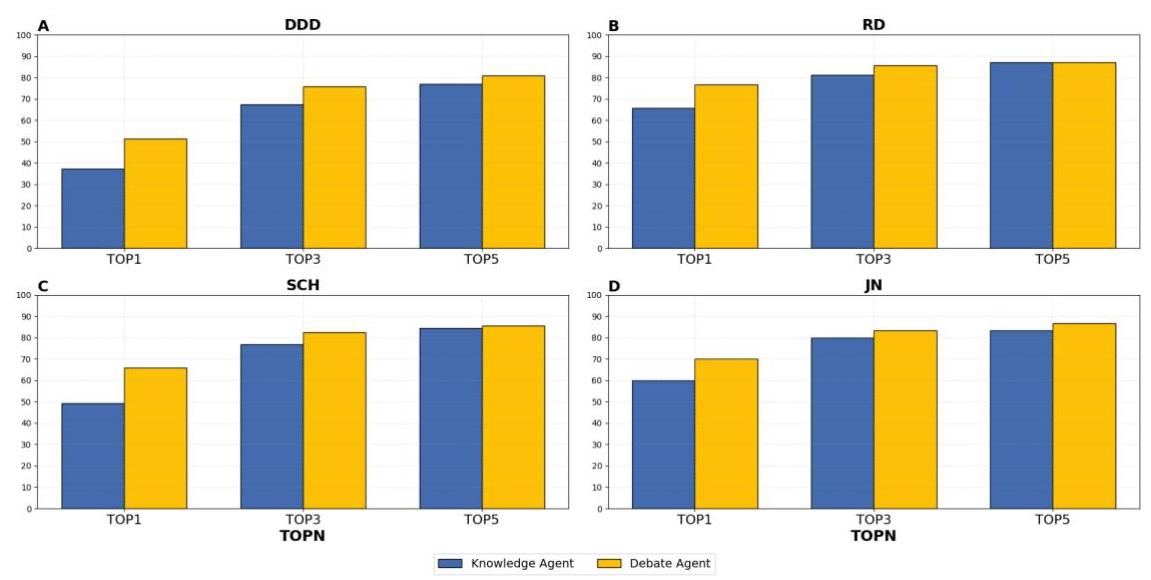
图3:知识智能体和辩论智能体在四个孟德尔遗传疾病数据集上的表现。该图显示了知识智能体(黄色条形)和辩论智能体(蓝色条形)在四个数据集(标记为A(DDD,合成),B(RD,合成),C(SCH,真实),和D(JN,真实))上的诊断准确率。
为了更好地理解辩论如何影响性能,我们根据是否TOP1答案识别出致病基因,将智能体响应分为八种不同的模式。每种模式由知识、数据和辩论智能体的响应序列表示,其中 ‘A’ 表示致病基因排名第一,‘B’ 表示不是。分类包括一致答案(所有三个智能体一致,标记为 A / A / A \mathrm{A} / \mathrm{A} / \mathrm{A} A/A/A 或 B / B / B \mathrm{B} / \mathrm{B} / \mathrm{B} B/B/B )、召回答案(知识或数据智能体未将致病基因列为TOP1,但辩论智能体列为TOP1,如 B/B/A, A/B/A 和 B/A/A),以及遗漏答案(至少有一个知识或数据智能体将致病基因列为TOP1,但辩论智能体未列为TOP1,如 B/A/B, A/B/B 和 A/A/B)。
这种结构化分析提供了对诊断过程动态的关键洞察。如图4A所示,统计结果显示所有数据集的一致性率超过 60 % 60 \% 60%,RD数据集的一致性率最高为 75 % 75 \% 75%,DDD数据集的一致性率最低为 62.4 % 62.4 \% 62.4%。高一致性率表明智能体之间有强烈的协议,特别是在 B / B / B \mathrm{B} / \mathrm{B} / \mathrm{B} B/B/B 场景中,没有一个智能体将致病基因列为TOP1,这表明尽管结果为负面,但在诊断观点上达成了一致。
此外,在DDD、RD和SCH数据集中,召回率始终超过遗漏率,表明辩论智能体通过重新校准初始差异有效提升了诊断准确性。JN数据集展现了最高的召回率
30
%
30 \%
30%,同时
保持零遗漏率,表明即使在表型较少的情况下,如侏儒症(图4),多智能体辩论依然有效。这些发现强调了辩论智能体不仅确认正确的初步诊断,还能有效纠正错误诊断的关键作用。在其他数据集中,辩论智能体成功纠正了知识和数据智能体的不准确之处(类别B/B/A),通过结构化辩论动态改进诊断评估的能力,增强了多智能体系统实现更高诊断准确率和发现现有知识之外的新医学洞见的潜力。
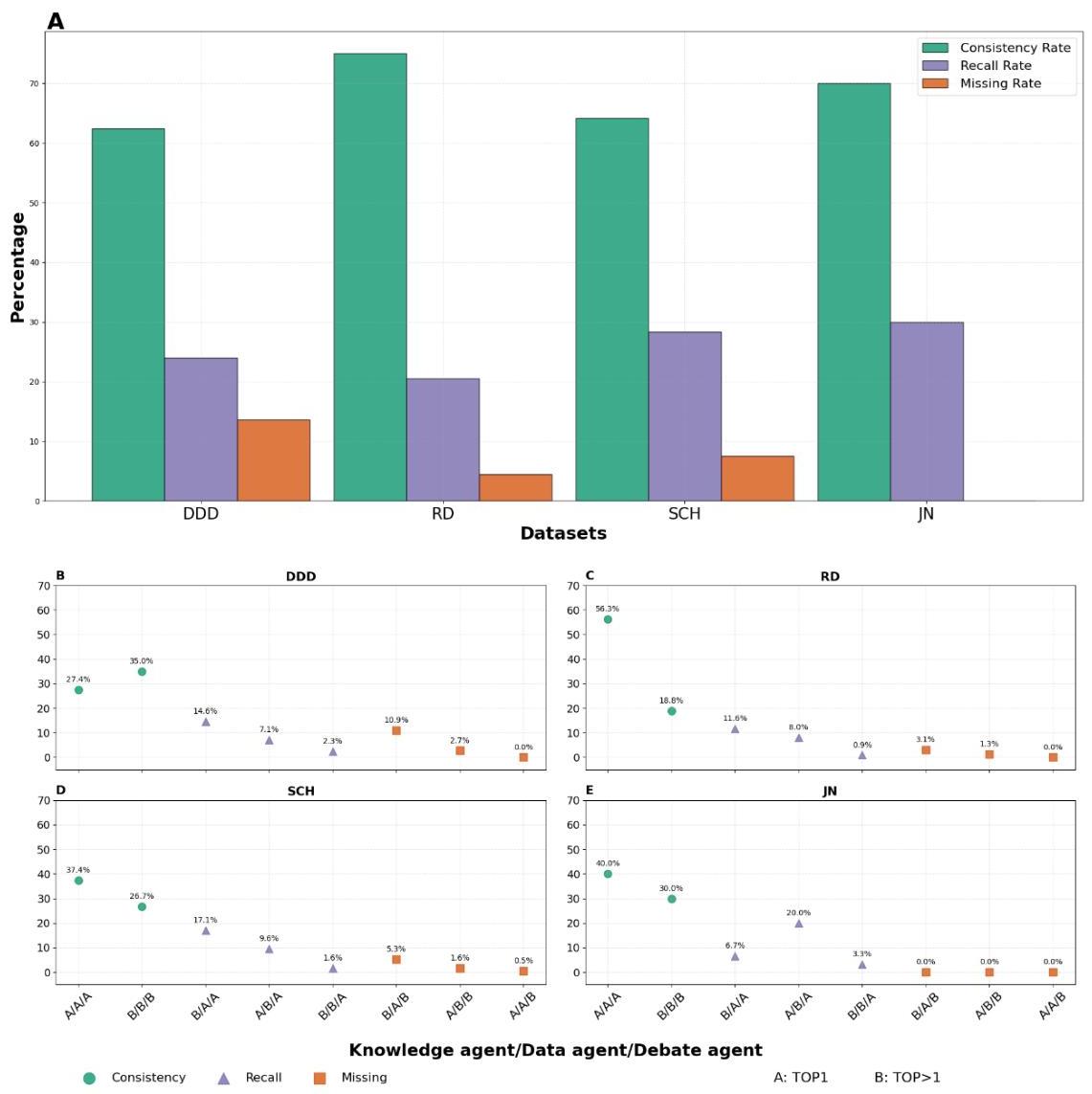
图4:MD2GPS在四个数据集中的响应模式表现。A)四个数据集中一致性、召回率和遗漏率的汇总。BCDE)统计分析将多智能体响应分为八个组,每组包含知识、数据和辩论智能体的顺序响应。在该系统中,‘A’ 表示致病基因排名首位,而 ‘B’ 表示不是。绿色圆圈代表一致答案,紫色三角形表示召回答案,橙色矩形表示遗漏答案。
为进一步展示MD2GPS的功能,我们讨论了一个B/B/A的例子。在这个例子中,知识智能体、数据智能体和辩论智能体分别提出PEX6、HLADRB1和TBC1D24作为致病基因。他们从各自独特的分析角度评估患者的表型。
具体来说,知识智能体强调听力损伤和手部异常为主要表型。相比之下,数据智能体通过其分析计算,优先考虑发热为主要表型。经过全面辩论后,辩论智能体调整了早期评估,确定癫痫是患者的主要且最严重的表型。这导致认识到TBC1D24,最初由辩论智能体排在首位,是与患者呈现的神经系统疾病最密切相关的基因。这个案例突显了MD2GPS通过其多智能体辩论系统整合多样诊断视角并细化基因-表型相关性的能力。这个案例展示了MD2GPS系统促进的细致决策过程,展示了其整合多样诊断见解并通过辩论细化最终基因-表型关联的能力。
评估MD2GPS在额外真实世界数据集中的扩展性能和临床潜力
真实世界数据集
我们在济宁医学院附属医院进行了MD2GPS系统的前瞻性临床应用,涉及320名被诊断为侏儒症的患者。最初,医生根据ACMG标准为识别的突变建立证据水平,确认72个为致病或可能致病。MD2GPS的辩论功能随后促进了进一步分析,结合文献咨询,确定了12名患者的致病基因。值得注意的是,整个诊断过程仅用了11天就完成了320名患者的诊断,相比三年前首次诊断所需的四个月时间减少了90%。在这些病例中,MD2GPS的TOP1和TOP5准确率分别为66.7%和100%,显著优于其他系统如Xrare和diseaseGPS。
该研究还发现了60个病例中识别的突变与观察到的表型不一致的情况,表明可能存在需要进一步探索的多基因相互作用。这些发现不仅证明了MD2GPS在临床环境中的效率和准确性,还展示了其挖掘新的医学研究途径的能力,强调了该系统扩大现有医学知识的实质性潜力。
讨论
在这项研究中,我们介绍了MD2GPS,这是一个创新的LLM驱动多智能体辩论系统,旨在提高遗传疾病诊断的可靠性和效率。与传统的Xrare和AMELIE模型不同,这些模型需要大量的训练并且在较少的罕见遗传疾病样本中可能会出现过拟合,MD2GPS无需特定的训练要求,并在TOP1得分上平均提高了 23.1 % 23.1 \% 23.1%,从 42.9 % 42.9 \% 42.9% 提高到 66 % 66 \% 66%。在临床环境中,MD2GPS不仅提高了诊断的精确度,还显著将诊断时间缩短了大约 90 % 90 \% 90%,极大地简化了遗传诊断过程。
为了适应持续的技术进步,MD2GPS被设计为模块化架构,允许对关键组件进行轻松更新。这种设计允许无缝替换数据智能体中的排名算法,以及知识和辩论智能体中的LLMs和提示。这种灵活性确保MD2GPS始终处于遗传诊断的前沿,能够快速适应新兴技术。
MD2GPS主要针对涉及SNV和Indel变异的孟德尔疾病样本开发和评估,表现出色。然而,MD2GPS还能够处理其他类型的遗传变异,包括非编码变异、结构变异和拷贝数变异(CNVs)。它允许将这些新型变异类型集成到数据智能体中,使用户可以直接输入其结果或结合自己的生物信息学管道。MD2GPS的主要限制在于数据智能体目前仅支持单样本分析,而不支持基于家族的样本分析。然而,用户仍然可以将家族样本分析的结果输入到数据智能体中进行后续辩论和进一步分析。
此外,虽然MD2GPS旨在诊断遗传疾病,但多智能体辩论框架也适用于其他复杂病症的诊断,例如
阿尔茨海默病和帕金森病。这些神经退行性疾病是多因素的,涉及遗传易感性和环境因素之间的复杂相互作用。通过将识别与这些疾病相关的遗传标记的算法集成到数据智能体中,MD2GPS可以提供初步的遗传评估。此外,知识智能体可以根据最新关于生活方式和环境因素对疾病进展影响的研究进行定制,包括相关提示。在辩论期间,智能体可以评估和讨论各种遗传和非遗传因素的重要性及影响,提供一个整合当前分析和现有知识的全面视图。
MD2GPS不仅对潜在的致病基因进行排名,还阐明了其排名的原因,解释为什么某个特定的遗传变异被排在首位而不是其他。这种可解释性对于分析多基因效应至关重要,对理解复杂的遗传相互作用具有重要意义。这一点在一名六岁被诊断为努南综合征的病例中得到了体现,MD2GPS与临床顶级诊断一致,即KRAS p.V14I突变,并同时指出MTOR突变为同等致病性。这一见解暗示了多基因效应,可能加剧患者的症状,如颈部蹼状现象——这是单独KRAS p.V14I突变通常不会引起的现象。通过揭示传统模型可能忽略的复杂遗传关系,MD2GPS提供了更深的洞察,并有助于制定更具针对性的治疗策略。
总之,MD2GPS通过其以LLM驱动、辩论为核心的途径彻底改变了孟德尔疾病的诊断。通过利用多智能体系统,它不仅超越了现有的诊断方法,还提供了一个更加动态和可扩展的平台,用于不断改进和应用遗传诊断。该系统不仅提高了诊断遗传疾病的速度和准确性,还在推进我们对遗传病理学及其治疗选择的理解中发挥了关键作用。
在线方法
数据准备
本研究利用了来自四个不同数据集的1,257个遗传疾病样本:两个真实数据集来自上海儿童医院(定义为SCH数据集)和济宁医学院
医院(定义为JN数据集),以及两个基于DDD数据库(定义为DDD数据集)或文献中的病例报告(定义为RD数据集)构建的数据集。
SCH数据集包括在上海儿童医院诊断为遗传疾病的187名患者,这些数据曾在我们的先前研究中使用 20 { }^{20} 20。它涵盖了326种疾病类别、超过380种异常表型和140种不同致病基因。所有患者都是基层医生难以诊断的病例,最终通过多学科团队会诊确诊。
JN数据集包括来自济宁医学院附属医院的102名患者,这些数据尚未被提出。它包括主要表现为身材矮小的患者,无显著的身体体征或生理生化检测异常。其中,30例被确认为具有可识别的致病基因,涉及12种异常表型和20种不同的致病基因。其余72个样本,根据ACMG指南被认为携带致病或可能致病的变异,但尚无明确的致病基因,表现出一致的临床表型:垂体性侏儒症(HP:0000839)、宫内生长迟缓(HP:0001511)、严重身材矮小(HP:0003510)和身材矮小(HP:0004322)。除此之外,未观察到其他具体的异常表型。
DDD数据集包含了来自解码发育障碍项目中的表型和基因型数据
30
{ }^{30}
30。最初,通过OMIM数据库筛选了6,085个病例,识别出基于致病基因及其遗传方式的确切遗传疾病。随后,为确保多样性和非冗余性,我们从每组具有相同致病基因的病例中随机选取一个样本,得到734个样本的子集。每个病例中的变异位点随后被随机分配到157个健康个体基因组中的一个,这些基因组作为背景基因组。这157个健康基因组由上海儿童医院提供,与DDD研究无关。该数据集涵盖了1242种疾病、超过1390种异常表型和734种不同的致病基因。
RD数据集是基于中国首批官方罕见病名单开发的,其中包括121种不同疾病,每种疾病都有既定的诊断和治疗方案
31
{ }^{31}
31。其中,根据特定标准排除了26种疾病:缺乏足够的病例研究报告,由体细胞或线粒体DNA变异引起的疾病。剩余的95种疾病,每种疾病由两到三个病例代表,共贡献了234个样本。表型数据和变异描述手动从公共文献中提取并总结。遗传信息,包括基因名称、cDNA突变及相应的蛋白质变化,使用TransVar
32
{ }^{32}
32转换为基因组DNA(gDNA)坐标,并在染色体上进行左对齐标准化。原始的自然语言描述首先使用PhenoGPT
33
{ }^{33}
33转换为HPO术语,然后由两位经验丰富的专家仔细审查和优化。因此,RD数据集现在包含95种疾病、超过870种异常表型和144种不同的致病基因。
数据智能体
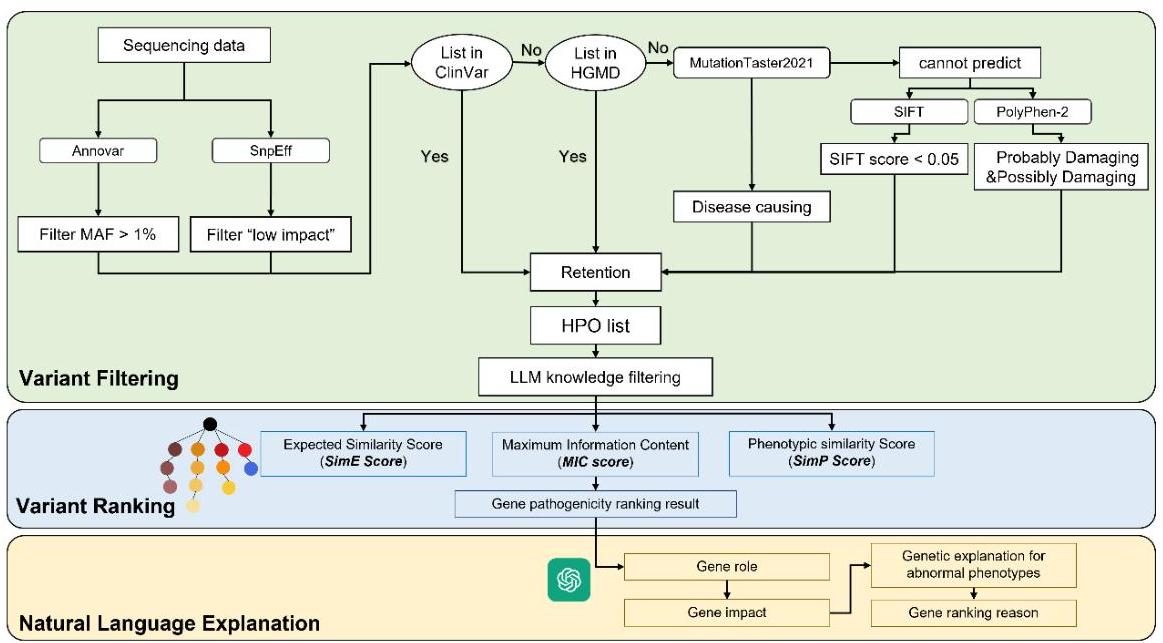
图5:数据智能体的功能架构,整合生物信息学数据分析与LLM解释。数据智能体整合了三个模块:变异过滤、变异排名和自然语言解释。通过生物信息学和基于表型的GPT-4过滤器精炼的基因数据,使用集成算法进行排名。然后GPT-4生成这些排名结果的结构化自然语言解释。MD2GPS中的数据智能体由三个主要模块组成:变异过滤、变异排名和自然语言解释(图5)。在变异过滤过程中,使用ANNOVAR
34
{ }^{34}
34和snpEff
35
{ }^{35}
35对变异呼叫格式(VCF)中的遗传变异进行注释,
提供全面的细节,如基因位置、氨基酸变化、人群中等位基因频率、基于数据的变异影响预测以及来自HGMD
36
{ }^{36}
36和ClinVar
37
{ }^{37}
37等数据库的注释。然后,分阶段过滤注释的变异,首先排除在普通人群中次要等位基因频率(MAF)大于
1
%
1 \%
1%的变异。随后,排除在ClinVar数据库中分类为"良性"或"可能良性"的变异。未被ClinVar覆盖或标记为"不确定"的变异,如果在HGMD中记录为致病性,则予以保留。进一步通过剔除MutationTaster2021
38
{ }^{38}
38预测为’多态性’的变异进行精炼。对于无法通过MutationTaster2021评估的变异,剔除SIFT评分
39
{ }^{39}
39大于等于0.05且PolyPhen-2
40
{ }^{40}
40预测为非致病性的变异。最后,使用集成的GPT-4模型评估每个基因的功能损害与患者至少一种表型的相关性,排除与表型完全无关的基因。
在变异排名过程中,计算三个关键指标对变异进行评分:疾病经验相似性得分(SimE)、临床表型最大信息含量(MIC)和疾病理论相似性得分(SimT),后者聚合了基于表型和基于基因型的疾病相似性得分。基于这些指标,所有潜在的遗传疾病按照优先级降序排列,以便有条理地识别最可能影响患者的遗传条件。
最后,数据智能体利用GPT-4根据排名结果生成自然语言解释。这包括阐述基因功能、详细说明基因与疾病之间的分子机制联系,以及解释与疾病相关的基因致病性排名的原因。这一叙述层不仅有助于理解计算结果,还增强了向临床从业者和研究人员传达发现的能力,确保清晰和可操作的见解。
知识智能体
在MD2GPS框架内,知识智能体使用GPT-4模型处理数据智能体提供的无序突变过滤和排名结果。这允许对基因突变对疾病的贡献进行全面分析,接受诸如症状列表和自然语言表述的基因突变等输入。
在分析过程中,每个基因突变的作用、对患者生理的影响以及排名理由都被仔细探讨,以澄清其在疾病中的致病作用。特别强调了选择特定基因序列背后的逻辑——不仅基于每个基因的生物学功能和对症状的贡献,还要考虑基因间的相互作用。这种详尽的分析,与对比其他潜在序列相对照,解释了为何所选序列更能准确反映患者的实际病情。
知识智能体的成果分为两部分:排名以JSON格式输出,以确保数据透明度并方便后续分析,而推理解释则以自然语言提供,为诊断过程提供科学和逻辑基础。
多智能体辩论
MD2GPS中的辩论智能体通过整合知识和数据智能体的响应进行辩论,这些响应包括变异排名、解释和来自ClinVar等数据库的证据。使用GPT-4,代理通过结构化的提示引导基因致病性的辩论。输入包括基于生物信息学排名算法识别的前20个基因去重和随机化后的致病基因列表,格式化以应对GPT模型上下文长度的限制。每个提示引导GPT-4评估基因组合的系统效应,而不仅仅是单个基因的致病性。
在实施辩论过程中,辩论智能体评估知识智能体和数据智能体提供的证据的一致性和准确性,确保所依赖的信息与当前的遗传知识相符。此外,它还对严重性进行严格评估,分析每个表型的即时和长期影响,并识别直接由遗传变异引起的表型,以确定哪些基因突变对疾病进展最为关键。在基因排名过程中,优先考虑影响患者表型并被归类为高可信度变异的基因,确保重点放在最可能导致疾病的基因上,从而提高诊断的特异性和效率。
此外,辩论智能体强调因果关系的清晰性,要求在推断基因突变对疾病的影响时清楚界定直接因果路径。
这种因果关系的清晰性有助于更深入地理解疾病机制,并支持更有效的治疗策略的发展。考虑到疾病的多基因性质,辩论智能体还全面评估基因之间的相互作用及其对疾病表型的集体贡献。这种全面分析反映了疾病的复杂性,需要多维度的方法来解释遗传数据,从而增强对疾病遗传背景和进展的整体理解。总的来说,这些过程如下公式化:
数据智能体:
LLM { Gene 1 , Gene 2 , ⋯ } ∈ S gene ⋅ [ H P O 1 , H P O 2 , ⋯ } ∈ S H P O ⋅ R Gene ranks ] R Data Agent R Data Analysis 知识智能体: \begin{aligned} & \text { LLM } \\ & \left.\left\{\text { Gene }_{1}, \text { Gene }_{2}, \cdots\right\} \in \mathbb{S}_{\text {gene }} \cdot\left[H P O_{1}, H P O_{2}, \cdots\right\} \in \mathbb{S}_{H P O} \cdot \mathbb{R}_{\text {Gene ranks }}\right] \mathbb{R}_{\text {Data Agent }} \mathbb{R}_{\text {Data Analysis }} \\ & \text { 知识智能体: } \end{aligned} LLM { Gene 1, Gene 2,⋯}∈Sgene ⋅[HPO1,HPO2,⋯}∈SHPO⋅RGene ranks ]RData Agent RData Analysis 知识智能体:
辩论智能体:
S
gene
⋅
S
H
P
O
⋅
Z
Knowledge Analysis
Z
Data Analysis
⋅
S
ClinVar
[
R
Debate Agent
Z
Debate Analysis
]
\mathbb{S}_{\text {gene } \cdot \mathbb{S}_{H P O} \cdot \mathbb{Z}_{\text {Knowledge Analysis }}} \mathbb{Z}_{\text {Data Analysis } \cdot \mathbb{S}_{\text {ClinVar }}}\left[\mathbb{R}_{\text {Debate Agent }} \mathbb{Z}_{\text {Debate Analysis }}\right]
Sgene ⋅SHPO⋅ZKnowledge Analysis ZData Analysis ⋅SClinVar [RDebate Agent ZDebate Analysis ]
S
gene
\mathbb{S}_{\text {gene }}
Sgene 和
S
H
P
O
\mathbb{S}_{H P O}
SHPO 在患者的医疗记录中分别代表候选基因列表和异常表型集合。知识智能体根据医疗数据进行分析(
Z
Knowledge Analysis
\mathbb{Z}_{\text {Knowledge Analysis }}
ZKnowledge Analysis ),而数据智能体利用医疗数据及其自身计算的基因排名(
R
Gene ranks
\mathbb{R}_{\text {Gene ranks }}
RGene ranks )进行分析(
Z
Data Analysis
\mathbb{Z}_{\text {Data Analysis }}
ZData Analysis ),两者均由LLMs驱动。随后,辩论智能体整合了两个智能体生成的分析和数据智能体提供的致病证据ClinVar(
S
ClinVar
\mathbb{S}_{\text {ClinVar }}
SClinVar ),进行辩论并得出
Z
Debate Analysis
\mathbb{Z}_{\text {Debate Analysis }}
ZDebate Analysis 。
评估
在评估MD2GPS框架在变异优先级排序中的有效性时,采用了一种系统的方法,使用TOPN累积分布函数(CDF)指标-TOP1、TOP3和TOP5。这些指标评估致病基因在前N预测中的可能性,反映在每个特定排名之前准确识别致病基因的病例累计百分比,确保对算法性能进行公平和全面的比较。"一致答案"定义为所有智能体-知识、数据和辩论-对基因的致病状态一致同意的情况,无论是将其排名为TOP1还是根本不排名。"召回答案"是指在初始存在差异后,通过辩论审议使致病基因升至TOP1的情况,而"遗漏答案"是在辩论后基因从TOP1下降的情况。"一致性率"通过一致答案的百分比测量,"遗漏率"通过遗漏答案的频率测量,"召回率"计算为召回答案占总病例的比例,突出系统纠正初始不准确性的能力。在济宁医学院附属医院诊断为侏儒症的队列中特别监测了诊断时间,以评估诊断过程在实际临床环境中的影响和速度。
参考文献
- Church, G. Compelling Reasons for Repairing Human Germlines. New England Journal of Medicine 377, 1909-1911 (2017).
- Posey, J.E., et al. Insights into genetics, human biology and disease gleaned from family based genomic studies. Genetics in Medicine 21, 798-812 (2019).
- Rahit, K.T.H. & Tarailo-Graovac, M. Genetic modifiers and rare mendelian disease. Genes 11, 239 (2020).
- Amberger, J.S., Bocchini, C.A., Scott, A.F. & Hamosh, A. OMIM.org: leveraging knowledge across phenotype-gene relationships. Nucleic Acids Res 47, D1038-d1043 (2019).
- Boycott, K.M., Vanstone, M.R., Bulman, D.E. & MacKenzie, A.E. Rare-disease geneticsin the era of next-generation sequencing: discovery to translation. Nat Rev Genet 14, 681-691 (2013).
- Yang, Y., et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med 369, 1502-1511 (2013).
- Trujillo, D., et al. Clinical exome sequencing: results from 2819 samples reflecting 1000 families. Eur J Hum Genet 25, 176-182 (2017).
- Ewans, L.J., et al. Whole exome and genome sequencing in mendelian disorders: a diagnostic and health economic analysis. Eur J Hum Genet 30, 1121-1131 (2022).
- Lek, M., et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285-291 (2016).
- Stoller, J.K. The challenge of rare diseases. Chest 153, 1309-1314 (2018).
- Faviez, C., et al. Diagnosis support systems for rare diseases: a scoping review. Orphanet Journal of Rare Diseases 15(2020).
- Kay, V.J. & Irvine, D.S. Successful in-vitro fertilization pregnancy with spermatozoa from a patient with Kartagener’s syndrome: Case Report. Human Reproduction 15, 135-138 (2000).
- Khalifa, E., et al. Successful fertilization and pregnancy outcome in in-vitro fertilization using cryopreserved/thawed spermatozoa from patients with malignant diseases. Hum Reprod 7, 105-108 (1992).
- Pirtea, P., et al. Successful ART outcome in a woman with McCune-Albright syndrome: a case report and literature review. J Assist Reprod Genet 40, 1669-1675 (2023).
- Winkler, I., et al. A Successful New Case of Twin Pregnancy in a Patient with Swyer Syndrome-An Up-to-Date Review on the Incidence and Outcome of Twin/Multiple Gestations in the Pure 46,XY Gonadal Dysgenesis. Int J Environ Res Public Health 19(2022).
- Yu, Y., et al. Confrontment and solution to gonadotropin resistance and low oocyte retrieval in in vitro fertilization for type I BPES: a case series with review of literature. J Ovarian Res 14, 143 (2021).
- Smedley, D., et al. Next-generation diagnostics and disease-gene discovery with the Exomiser. Nat Protoc 10, 2004-2015 (2015).
- Robinson, P.N., et al. Interpretable Clinical Genomics with a Likelihood Ratio Paradigm. Am J Hum Genet 107, 403-417 (2020).
- Javed, A., Agrawal, S. & Ng, P.C. Phen-Gen: combining phenotype and genotype to analyze rare disorders. Nat Methods 11, 935-937 (2014).
- Huang, D., et al. diseaseGPS: auxiliary diagnostic system for genetic disorders based on genotype and phenotype. Bioinformatics 39(2023).
- Sifrim, A., et al. eXtasy: variant prioritization by genomic data fusion. Nat Methods 10, 1083-1084 (2013).
- Li, Q., Zhao, K., Bustamante, C.D., Ma, X. & Wong, W.H. Xrare: a machine learning method jointly modeling phenotypes and genetic evidence for rare disease diagnosis. Genet Med 21, 2126-2134 (2019).
- Birgmeier, J., et al. AMELIE speeds Mendelian diagnosis by matching patient phenotype and genotype to primary literature. Sci Transl Med 12(2020).
- Fouad, Y., et al. The NAFLD-MAFLD debate: eminence vs evidence. Liver International 41, 255-260 (2021).
- Garrett, M., Schoener, L. & Hood, L. Debate: A teaching strategy to improve verbal communication and critical-thinking skills. Nurse educator 21, 37-40 (1996).
- Rafailov, R., et al. Direct preference optimization: Your language model is secretly a reward model. Advances in Neural Information Processing Systems 36(2024).
- Lin, Z., Niu, Z., Wang, Z. & Xu, Y. Interpreting and Mitigating Hallucination in MLLMs through Multi-agent Debate. arXiv preprint arXiv:2407.20505 (2024).
- Wei, J., et al. Chain-of-thought prompting elicits reasoning in large language models. Advances in neural information processing systems 35, 24824-24837 (2022).
- Slonim, N., et al. An autonomous debating system. Nature 591, 379-384 (2021).
- Firth, H.V., et al. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am J Hum Genet 84, 524-533 (2009).
- Zhang, S. Compendium of China’s first list of rare diseases, (People’s Medical Publishing House, 2018).
- Zhou, W., et al. TransVar: a multilevel variant annotator for precision genomics. Nature Methods 12, 1002-1003 (2015).
- Yang, J., et al. Enhancing phenotype recognition in clinical notes using large language models: PhenoBCBERT and PhenoGPT. Patterns 5, 100887 (2024).
- Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic acids research 38, e164-e164 (2010).
- Cingolani, P., et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6, 80-92 (2012).
- Stenson, P.D., et al. Human Gene Mutation Database (HGMD): 2003 update. Hum Mutat 21, 577-581 (2003).
- Landrum, M.J., et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res 42, D980-985 (2014).
- Steinhaus, R., et al. MutationTaster2021. Nucleic Acids Research 49, W446-W451 (2021).
- Ng, P.C. & Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic acids research 31, 3812-3814 (2003).
- Adzhubei, I.A., et al. A method and server for predicting damaging missense mutations. Nat Methods 7, 248-249 (2010).
参考论文:https://arxiv.org/pdf/2504.07881



















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











