







作者: 吴炜坤
参考1: Protein structure determination using metagenome sequence data
参考2: Learning Generative Models for Protein Fold Families
参考3: http:// 2016.rosetta.ninja/day- 2/tutorial-protein-folding
一、前言
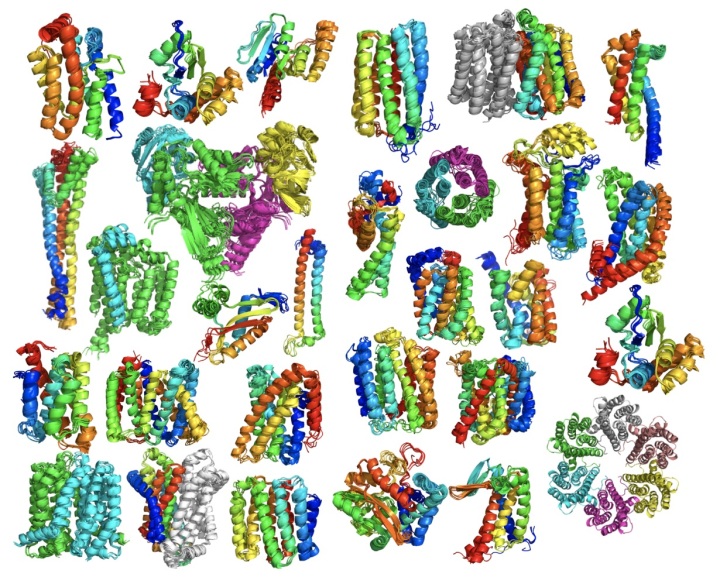
目前依然有大约5200个蛋白质家族缺少结构信息。在2017年,David Baker实验室成功利用宏基因组数据,对614个蛋白质家族进行三维建模,其中206个家族为膜蛋白,137个家族在PDB数据库中未见有代表性的结构。
目前已有58个家族的晶体被解析,通过与实验晶体比对,Rosetta+GREMLIN的+宏基因组数据的预测方法得到的结果整体精度还是相当的可观!利用共进化信息可以快速地预测蛋白质家族的代表性结构特征。
二、 计算原理
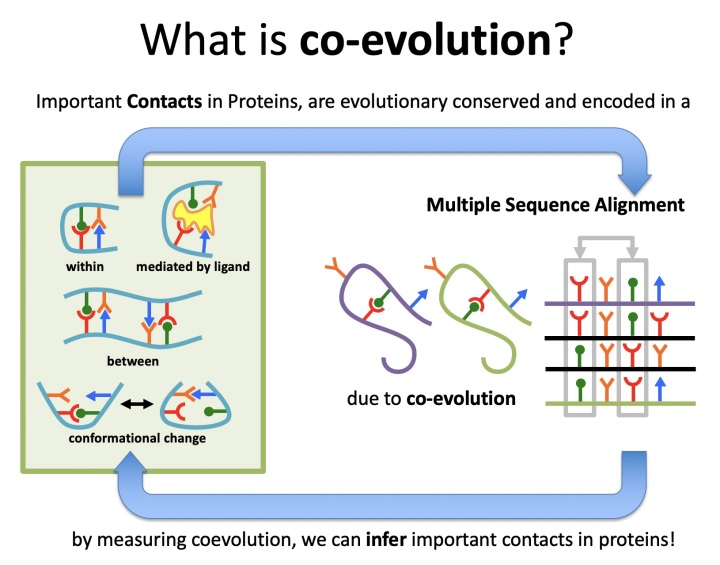
1 什么是蛋白质的共进化信息?
在生物进化过程中,蛋白质三维结构中与功能相关的重要区域的相互作用对往往是保守的,但是其氨基酸的空间位置是可以位置交换的。通过对序列进行比对,我们可以找到这些重要的残基对,并将这些信息利用到蛋白质3D建模中,对它们的距离进行限制。
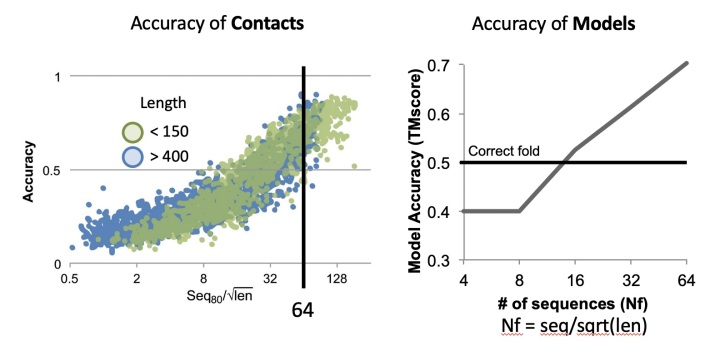
2 共进化信息的使用门槛
共进化的信息可以来源于宏基因组的数据。但是使用共进化信息到建模过程中是需要一定条件的,如果某个家族内的序列数目小于一定的阈值,那么从中提取的共进化信息可能是不够准确的。研究表明,家族内基因的effective number大于64时,对蛋白质相互作用分析较为准确,并且模型质量(TMscore可达0.7)
三、使用GREMLIN、 abinitio relax以及Hybridize进行同源模型构建
注: 由于计算方法比较复杂,此处没有整合宏基因组数据;详细参考文献原文。
1 使用GREMLIN生成共进化信息限制参数
预测的结构域最好在100个氨基酸左右,此处以赖氨酸tRNA合成酶C末端结构域为例:
>test
MQRQPVSSSRILSIGYDPDNRMLEIQFREQGTYQYLGVPERAHQNFMSAVSKGRF 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章

















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









