






《Chromatin state dynamics during blood formation》
虽然是2014年的science,但是数据非常好(小鼠全血,从HSC造血祖祖细胞,到髓系、淋系、红系的所有亚细胞)
对于构建血液分化过程中的转录组选择性剪切事件是非常好的研究模型;
那么废话不多说,我们选择这个14年的science,其中有183个(Chip-seq以及ATAC-seq的数据),52个RANseq的数据。我们分别用mm10(UCSC的基因组)对比。Chip-seq和ATAC-seq用chipseq pipeline中经典的bowtie2对比,RNAseq则用经典的二代比对方法STAR;
本文的关键就在chipseq的测序方法可以给我们提供更多的promoter、enhancer、TSS区域的信息;
整个基本的研究背景和蓝图:
hemocytes cells development:

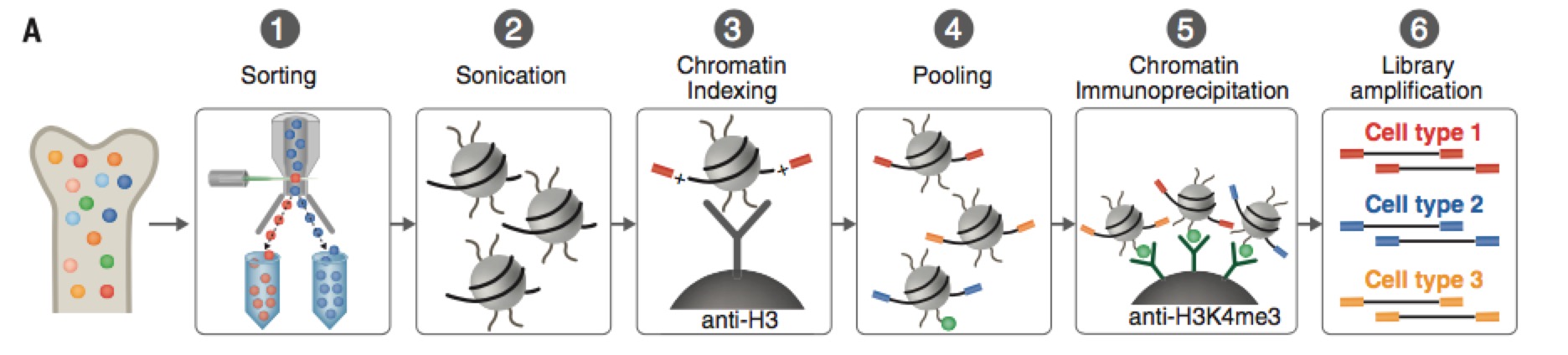
Chipseq library and seqencing:
文章一些比较founding的结果:
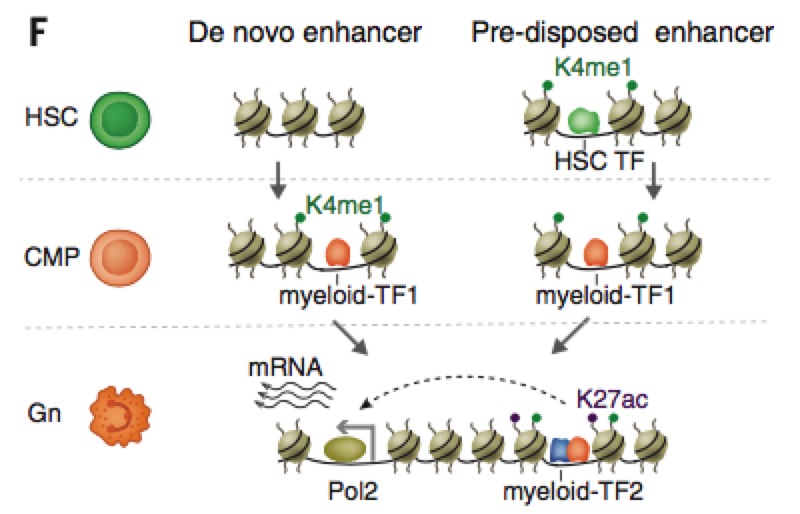
我们可以看到enhancer的分布是在血液细胞的不同分化时期有明显的差异分布(根据%以及ratio值):
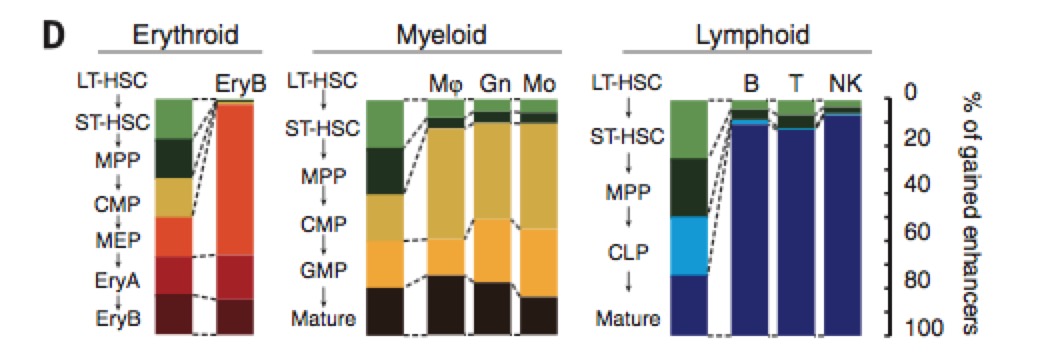
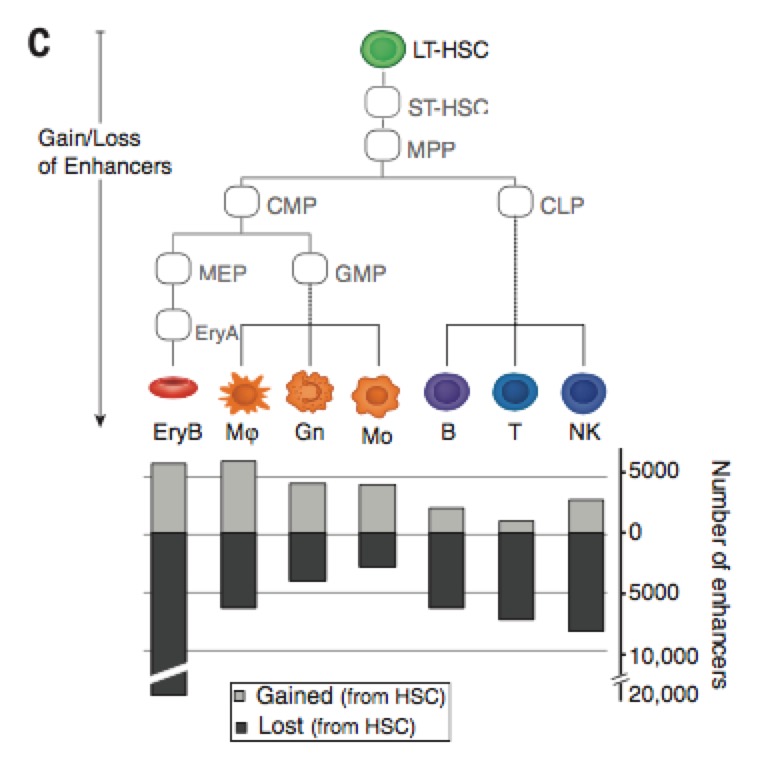
以及观察enhancer在不同分化时期的获得以及损失量:
跟RNAseq的数据做比较:
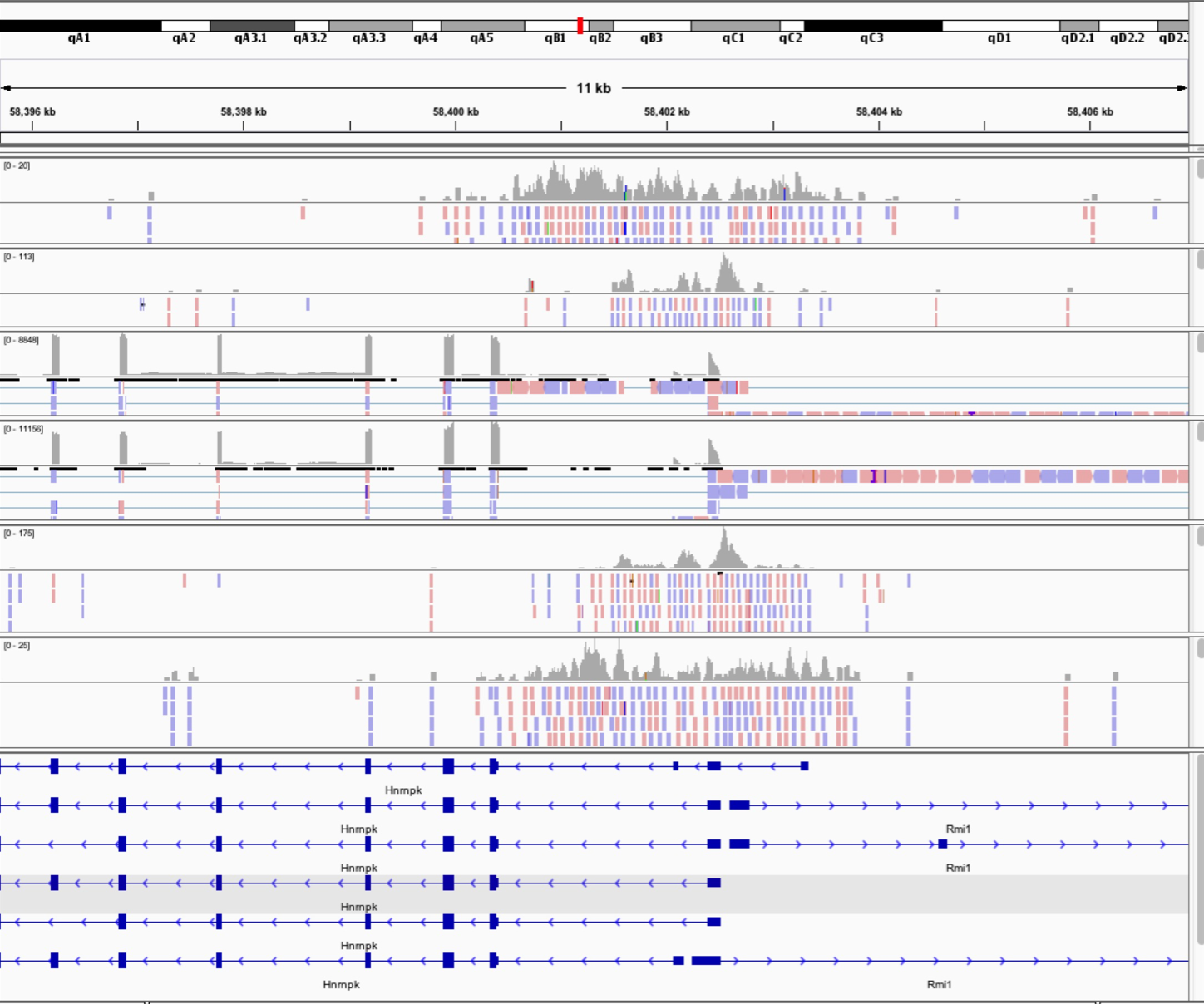
(这是本人的小鼠数据所分析出来的有first exon差异同一基因的转录本,可以看到的是在exon区域rnaseq数据都非常好的reads mapping上去,同时我们的CHIPseq以及ATACseq数据在TSS区域都有一个很好的富集以及peaks的覆盖,但是promoter以及TSS区域的peaks通常都比first exon的reads位置要前移一些,更好更直观的看出了chip call peaks 与纯二代测序出来的reads的差别,和可揭示的不同深度和程度的生物学问题)
那么在分析chipseq数据的时候我按照chipseq标准流程:
从下载的sra文件、fq文件(质控)、bowtie2比对、bam文件(bam-QC,check比对率)、MACS(call peaks)、CHIPseeker做peaks的可视化、或者是自己通过bioconductor上的TFBSTool去找相应的高表达的peaks以及regulatory binding sites;
用一张非常简单的chipseq pipeline的示意图来展示一下基本思路:
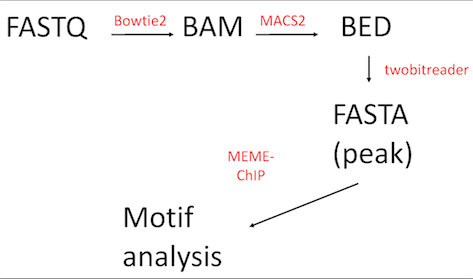
代码展示如下:
#####将SRA 文件转化为fastq文件
for i in $(ls SRR*)
do
/usr/bin/fasterq-dump --split-3 -O ./raw_data/ $i
done
#########对fq文件进行QC
for i in $(ls *fastq)
do
/usr/local/bin/fastqc -f fastq -o ./qc_files/ $i
done
######对fastq文件用 bowtie2 进行比对,生成bam files(注意bowtie以及bowtie2的区别,以及与STAR的使用情况的区别)
for i in $(ls *fastq)
do
/usr/bin/bowtie2 -p 6 -3 5 --local -x /reference/UCSC/mm10/bowtie2_index/mm10 -U $i | /usr/local/bin/samtools view -bS - | samtools sort - > /bam_files/$i\.bam
done
#####得到的bam文件进行sort以及index(by samtools)
####再用 bamtools 对相同 cell type 进行 merge 操作
/usr/bin/bamtools merge -list CLP_bamfiles.txt -out /merged_bam_files/CLP_merged.bam
#### 对merge好的文件去PCR重复
ls *merge.bam | while read id
do
nohup samtools markdup -r $id $(basename $id ".bam").rmdup.bam
done
#####bam to bed format
ls *rmdup.bam| while read id
do
bedtools bamtobed $id > $id ".bed"
done
###用MACS2 对bed 文件call peak(没有control sample)
ls *.bed | while read id
do
/usr/bin/macs2 callpeak -t $id -g mm --nomodel --shift 50 --extsize 100(reads length setting) -n ${id%%.*} --outdir ./callpeak_result/
done
######最后得到的文件就是
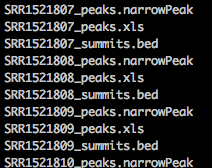
NAME_peaks.xls: 以表格形式存放peak信息,虽然后缀是xls,但其实能用文本编辑器打开,和bed格式类似,但是以1为基,而bed文件是以0为基.也就是说xls的坐标都要减一才是bed文件的坐标
NAME_peaks.narrowPeak:same as the format of NAME_peaks.xls, but the former is more suitable for R input
NAME_summits.bed:记录每个peak的peak summits,话句话说就是记录极值点的位置。MACS建议用该文件寻找结合位点的motif。能够直接载入UCSC browser,用其他软件分析时需要去掉第一行。
######再用bioconductor的ChIPseeker包进行下游的可视化操作:
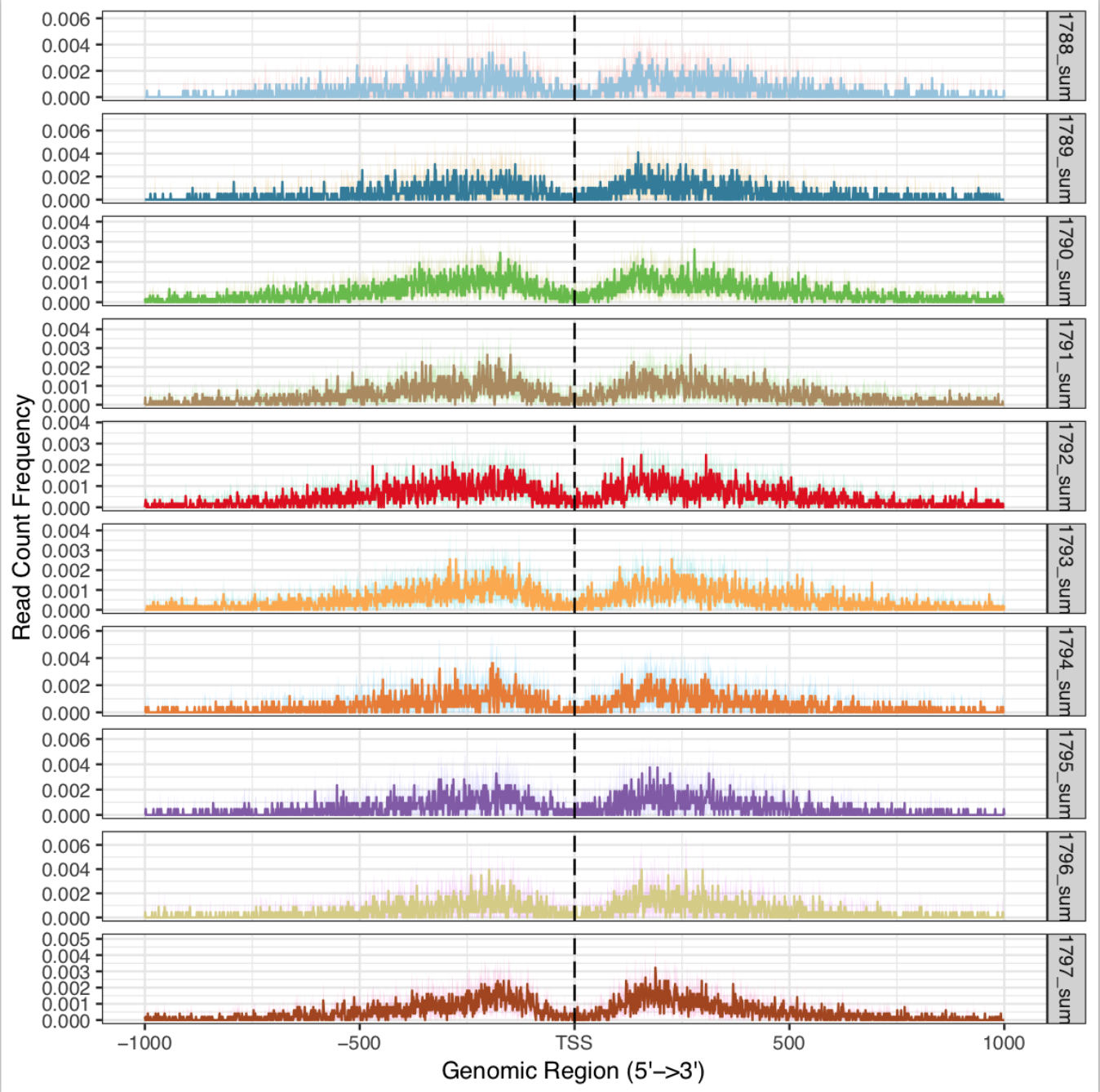
TSS附近区域的peak分布情况
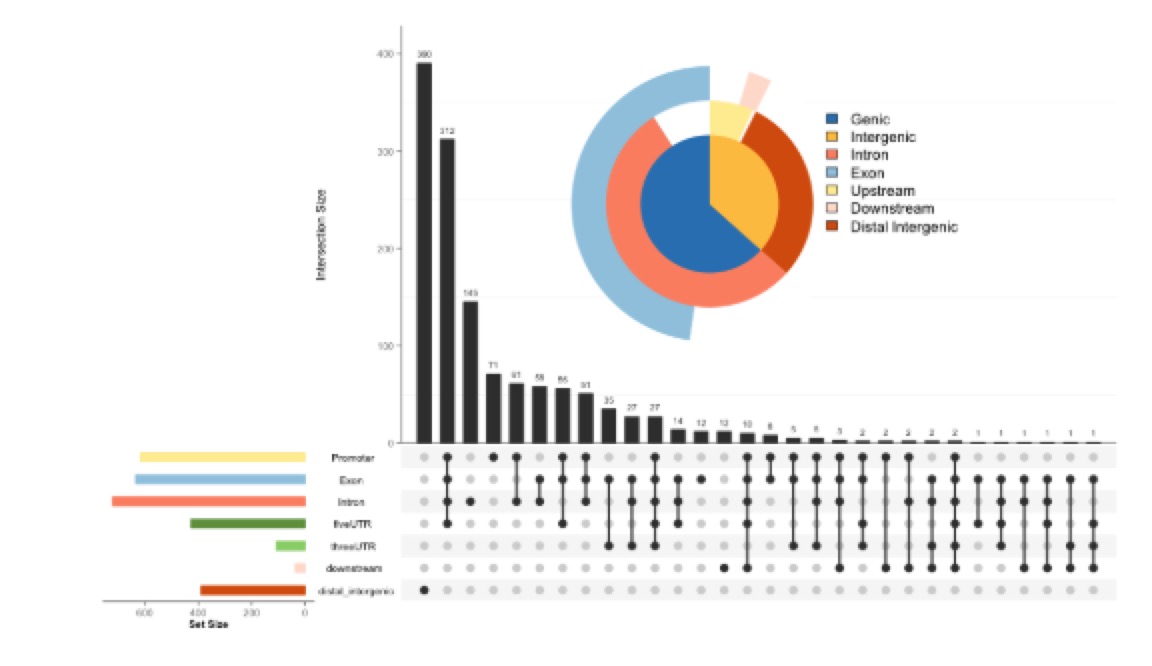
peak的组分分类upsetplot
用有peak annotations的基因进行GO、KEGG的富集分析:
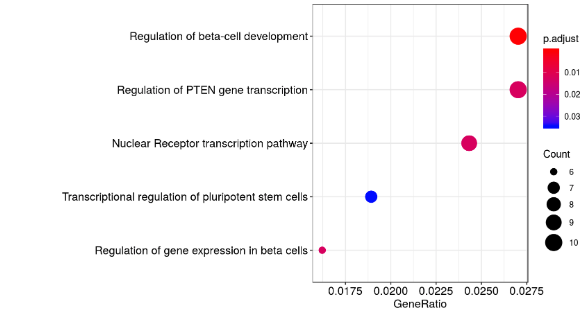
#非常常规的操作
那么我阐述了这么多之后,本随笔的观点主要是:
1、chipseq是很好的研究first exon、promoter、enhancer、TSS、TFbinding的好数据;
2、造血细胞的分化和发育是转录本水平研究的好模型;
3、TSS可以比较好的分出血细胞不同时期的亚型(不管是从PCA还是cluster)(后期会着重分享我们用TSS在不同cell type中的差异使用情况来对cell聚类分析,对TSS的ratio进行分类从而归纳出合适的差异使用程度分型);
4、人类的血液细胞和小鼠的血液细胞是都可以做TSS分析的,构建出来的TSS变化模型(pattern)是可以直接做物种间的保守性分析;
5、利用差异使用程度分型,找到cell specific的TSS事件,进行特定的motif富集以及注释;
4、AFE使用情况有差异的基因是可以进行基因功能富集以及基因之间调控网络的分析的;(主要是有GO、KEGG、付费的BioLayout Express3)
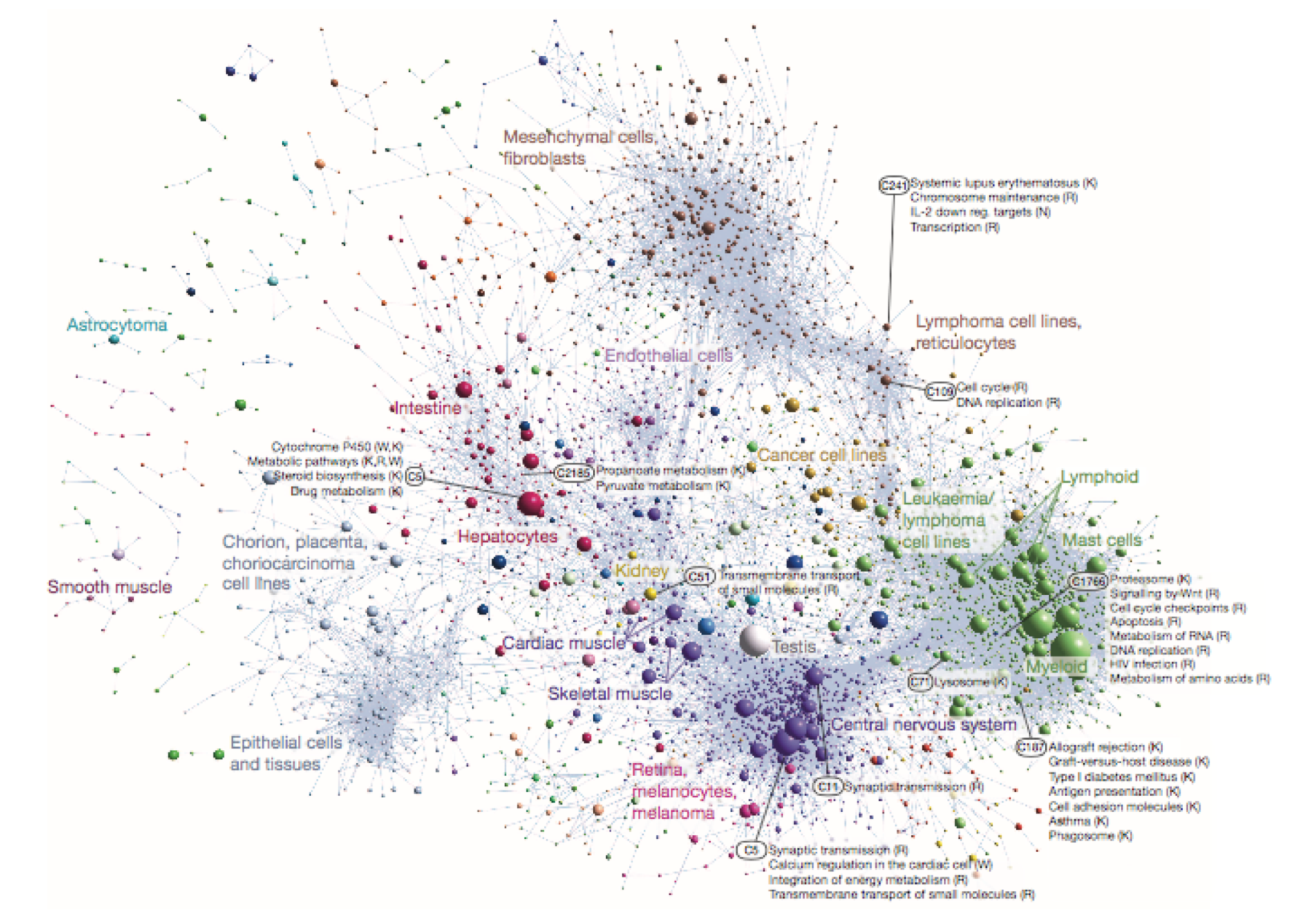 different AFE usage gene cluster
different AFE usage gene cluster
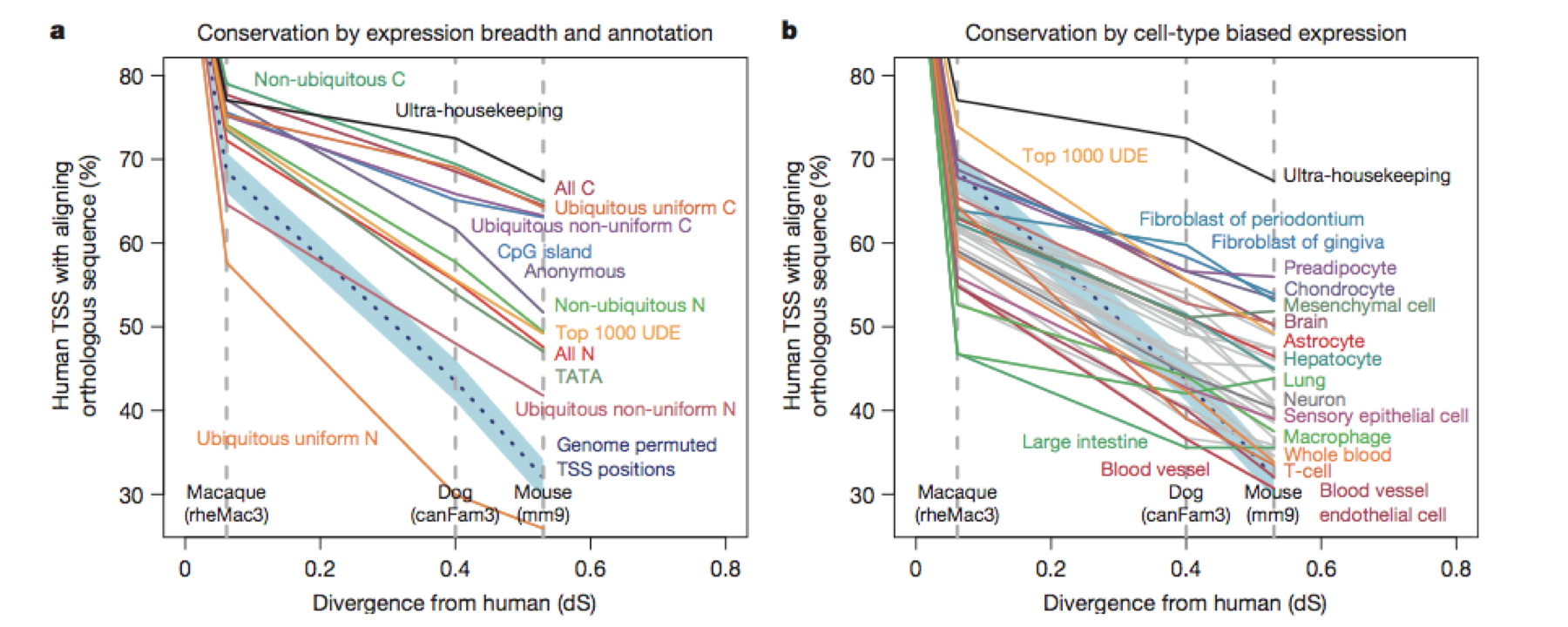 conservation between species
conservation between species
















 4952
4952











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









