






 本文深入探讨了分子轨道理论与价键理论的区别,通过实例解释了双原子和多原子分子的LCAO,并介绍了离域π键的π分子轨道分析。文中提出叠加/节点法,帮助理解分子轨道的成键性质,同时阐述了对称性匹配原则在分子轨道理论中的重要性。通过对苯、环辛四烯和茂等化合物的π系统分析,揭示了该方法在理解和预测分子轨道形状上的实用性。
本文深入探讨了分子轨道理论与价键理论的区别,通过实例解释了双原子和多原子分子的LCAO,并介绍了离域π键的π分子轨道分析。文中提出叠加/节点法,帮助理解分子轨道的成键性质,同时阐述了对称性匹配原则在分子轨道理论中的重要性。通过对苯、环辛四烯和茂等化合物的π系统分析,揭示了该方法在理解和预测分子轨道形状上的实用性。
本文版权归gfcjyb所有,转载请注明出处。
本文提供给中等、高级层级的无机、有机、结构化学学习者,为顺畅阅读本文,请确保已经熟练掌握了:价键理论,双原子分子的分子轨道理论,点群和特征标表。
目录
1 价键理论与分子轨道理论的区别
2 双原子分子和LCAO
3 多原子分子的LCAO与对称性匹配
4 离域π键的π分子轨道
5 环状离域π键的分析
1 价键理论与分子轨道理论的区别
在多原子分子中,价键理论会去分析比如这根σ键是谁的什么轨道和谁的什么轨道头碰头叠加形成的,而分子轨道理论认为每个轨道可以包含所有对称性匹配的原子轨道的贡献,实际上用量子化学算出来的每个轨道完全无法和单独的一根键去对应。
有些人可能会用一种杂糅了这两种理论的方式去理解。举个例子,水。按照这种杂糅理论,小明认为每个O-H单键会有一个O的sp3和H的1s叠加形成的σ成键轨道,然后还有一个对应的σ*轨道(反键轨道),然后O的两对孤对电子占据两个非键轨道。这样,小明觉得水的分子轨道的所有占据了电子的轨道就应该是(不考虑非价层的O的1s):两个简并的σ成键轨道,和两个简并的孤对电子非键轨道,后者应该是水的HOMO。
于是小明就去算了一下水的分子轨道,结果让他大跌眼镜:价层占据轨道的确是四个,但没有任何简并,四个轨道能量完全不同,而且HOMO看起来特别像水的pz,这不对啊?水不是sp2杂化啊?而且另外一对孤对电子去哪了?
好,我们现在就来分析小明的错误:分子轨道理论里是没有杂化的,杂化是纯粹的价键理论产物,那两对孤对电子同样也是。计算水的分子轨道时,可以理解为电脑根本不会认为O杂化了。
然后他的“两个轨道叠加形成一个成键轨道和一个反键轨道”想法也是错的,这只对双原子分子有效,他却认为在多原子分子中也是这样两两作用。这其实不能怪小明,只能怪小明的化学老师没解释清楚,或者怪现在很多大学化学课本对分子轨道理论就是讲到双原子分子为止,后面也不简单点一下,这样同学们再往后学离域π键分子轨道(高级的有机化学会碰到,尤其是周环反应)和“三个轨道叠加形成一个成键、一个反键和一个非键轨道”(高级的配位化学会碰到)时就会马上懵圈。
不过现在很多时候我们还是用价键理论解释很多事情的,比如带箭头的有机反应机理,以及一些键角等等。虽然现在也有一些用分子轨道去解释反应机理的例子(比如碱的HOMO与酸的LUMO叠加),但碰到HOMO是一大堆原子贡献的时候就不方便了。所以NBO诞生了,这个理论的核心是分子轨道部分定域化,所以挺像刚刚这个“杂糅理论”,但这就是另外一个故事了。
不过今天我们的主角是分子轨道,而我今天讲的主要是如何在已知计算结果的前提下用一种较易懂的方法去理解。这种方法其实在一定程度上会违背对称性,因此只能用来理解已知的结果,很多时候不能直接用来推断分子轨道的形状。这个道理大家要明白。
这个方法被我起名为叠加/节点法(Overlap/Node Method,简称OvNd),主要内容为:
分子轨道是满足一定对称性的原子轨道的线性叠加(这其实就是LCAO),相同符号的叠加会“融合”,不同符号叠加会“互斥”并在中间出现一个节点。融合会使能量降低,融合得越好(方向一致、大小近似等等)能量降得越低,反之亦然。
好了,核心说完了,接下来我们要先回到双原子分子这里。
2 双原子分子和LCAO
回到刚才的那条说法:“双原子分子中,两个轨道叠加形成一个成键轨道和一个反键轨道。”
那么我们用这个叠加/节点法来考虑一下这句话。大概就是下图这样(就以p轨道形成的σ、σ*和π、π*为例吧):
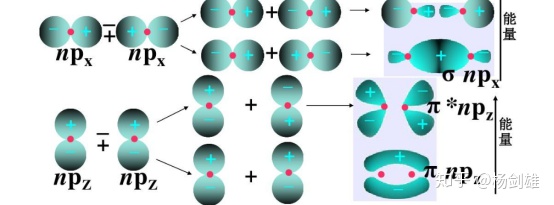
先看σ成键轨道。叠加前p轨道的排列让两个正号的波函数对在了一起,所以叠加后二者融合,按叠加/节点法,能量降低。类似地,反键轨道中二者排斥,在中间形成节点。这里说是排斥是因为可以看到中间的电子云明显比两侧小。
别忘了波函数本身没有物理意义,平方了才表示概率,波函数本身的正负号是纯数学正负,而刚刚说的叠加和排斥其实就是数学的带符号相加。两个正数加一起,肯定更正;一正一负碰一起,那不好说,要看哪边绝对值大,如果绝对值相同那就是0了。所以这就说明了为啥不同符号叠加后中间有节点。而为什么说“排斥”呢,因为不是0的部分因为这个叠加也减小了绝对值,一些绝对值小于了绘图阈限的位置(比如一个图里只画绝对值大于0.001的区域)就没了,所以看起来就特别像排斥。
这样上图的π和π*也可以类似地理解了吧。
3 多原子分子的LCAO与对称性匹配
那么回到刚才水的例子。现在理解了这些之后,就可以试着考虑水的价层成键轨道了。两个氢的1s不可能在O不参与的情况下直接叠加,因为太远了够不着。那么我们就可以分别考虑氧的2s、三个2p怎么样能和这两个氢叠加到一块去。
那么有人就要问了:为啥氧的这些原子轨道不会自己互相作用?好问题,用一句话回答就是:你把它们放在一起看看就知道了,根本没法叠加。标准的回答是:氧的这四个轨道对称性不同,所以无法叠加。
这就是对称性匹配。去看看C2v的特征标表,会发现px、py和pz的对称性都不同。s虽然和pz一样(注意:这里的z是C2轴方向,而非垂直于水分子平面的方向!)但pz的节点在中间呢,不可能和s叠加(不过反键轨道里这俩的确在一起了,所以σ反键轨道的情况要复杂很多,一般就是算一下看结果的事,这就是为啥我们现在只讨论成键轨道)。对称性匹配在这里的重要性在于,因为p轨道本身有对称性,因此和p轨道叠加的两个H的s轨道的组合必须表现出和这个p轨道一致的对称性。这条原则是分子轨道本身能“取消组合”(嗯就用这个词吧,熟悉ppt的小伙伴都知道我在说啥)成一些自己满足当前轨道的对称性的小组合时用到的。对称性匹配是分子轨道理论的核心原则之一,但和我的叠加/节点法没有必然联系,后面其实还会有冲突。
好了,回到水这里。既然明确氧的四个价层轨道必然存在且互相独立,那么我们就可以分别考虑每个轨道应该怎样和两个氢去叠加了。因为是成键轨道,所以要尽全力去叠加:
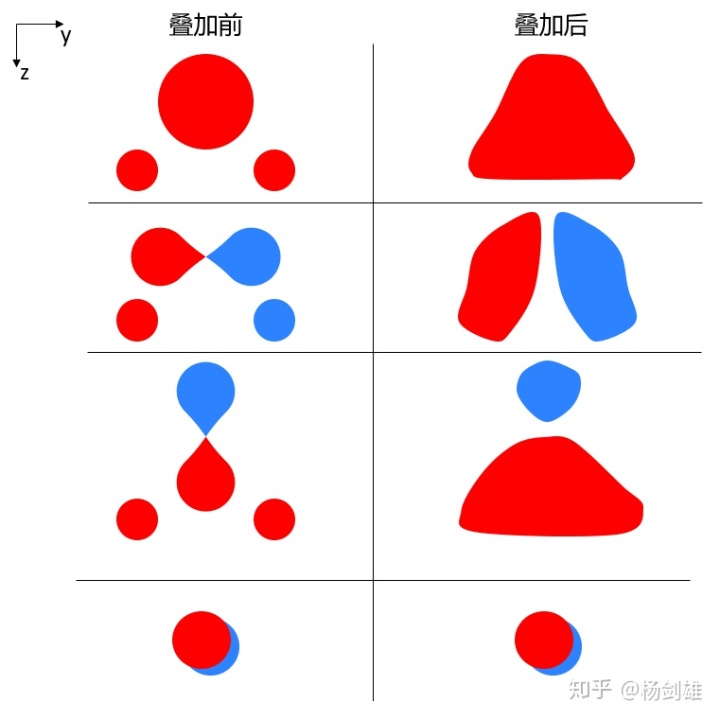
而这四个轨道的能量也应该是越来越高。这里O的py和H的s的叠加要弱于O的s和H的s,因为前者不是正碰(毕竟s球在球心连线方向怎么碰都是正碰)。而O的pz和H的s的叠加,因为p只有一半去和H的两个s叠加,而水的键角又是钝角,所以z方向上的叠加效果必然比y方向差。而px无法和H的s叠加,因为方向完全是不相干的,而yz平面上H面对的是px的节点,所以自然不能够叠加了。所以这个轨道按照分类完全可以算作是非键轨道。
之后我们算一下水的轨道,结果完全吻合。
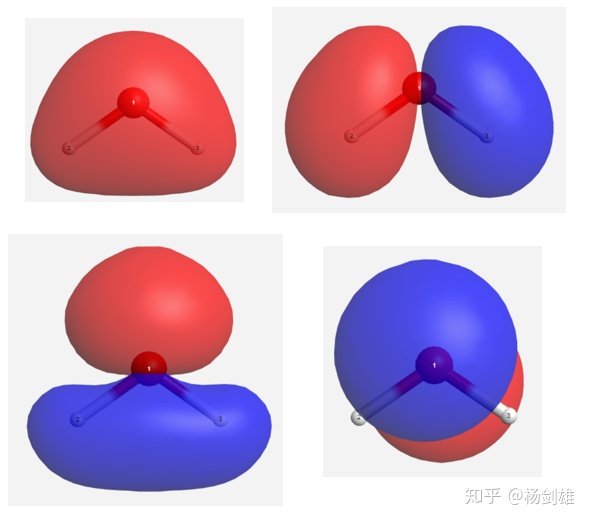
需要指出的是,这里我们并没有用上对称性,因为我们强调了作为填充轨道(或者说广义的成键轨道)必须尽全力去叠加,比如前三个轨道里你要是把其中一个H的符号(在这里表现为颜色)变一下的话就会立刻多出一个节点,这样其实对称性也没了。
那么我们再看一个例子:CH4。给大家一点时间用类似的办法想一想甲烷的价层成键轨道,一共是四个(按刚刚的杂糅理论那就是四个C-H σ键的成键轨道)。
思考时间剩余 10
思考时间剩余 9
思考时间剩余 8
思考时间剩余 7
思考时间剩余 6
思考时间剩余 5
思考时间剩余 4
思考时间剩余 3
思考时间剩余 2
思考时间剩余 1
好了,公布答案。这里我直接用周公度书里的图好了。
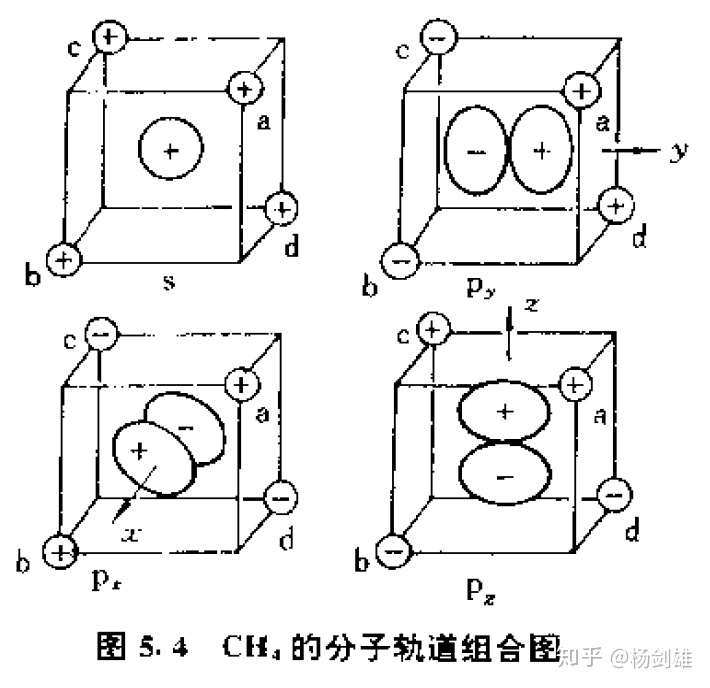
后三个轨道是简并的,因为叠加情况完全相同,只是方向不同而已。事实也的确如此。这里也没用到对称性,不过大家可以自己验证一下,四个H的s轨道组合表现出的对称性是和中心C原子的轨道一致的。
至于这些分子的未填充轨道,大家可以自己算算看看。LUMO和前几个也能用类似的办法解释,但毕竟出现节点的线性组合太多,所以到后面就真是相同对称性就揉在一起了(刚刚提到了,比如水的2s和2pz)。
4 离域π键的π分子轨道
好了,其实叠加/节点法应用的最好的地方就是离域π键的π分子轨道。为啥只是π分子轨道呢,因为π的成键轨道能量一般比σ高,反键又比σ低(原因是p肩并肩的叠加效果必然不如头碰头,这个很好理解),所以HOMO和LUMO一般都是它们,在研究周环反应、配位反应的机理中是非常重要的。而且因为pz(这里z就指垂直于分子平面的方向了)特殊的对称性,不会和σ搅在一起,所以π分子轨道不会有任何σ方面的贡献,所以研究它们只需要看这些pz就行了。
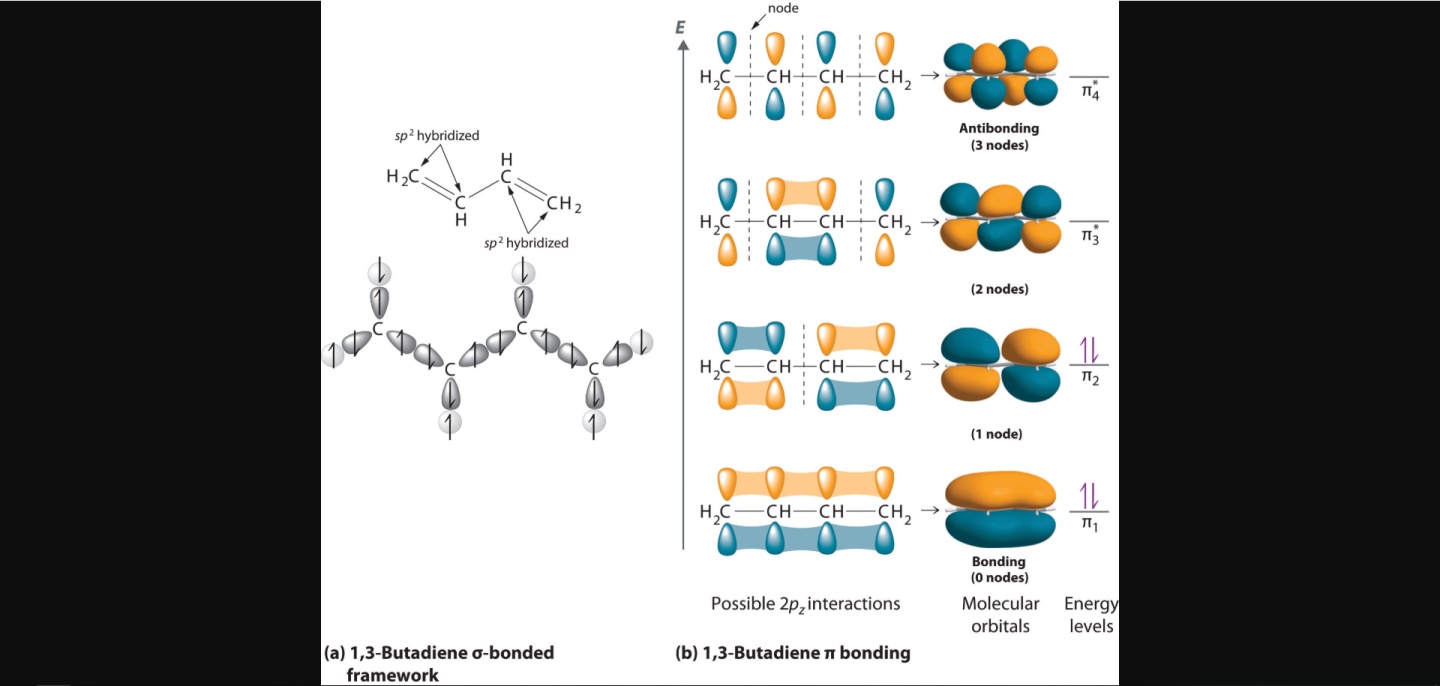
先看一个最经典的例子:1,3-丁二烯。
这里就是只考虑四个碳的pz的作用了,但是它们是一起作用的(所以这里就不能有前面的“两个轨道叠加形成一个成键轨道和一个反键轨道”的想法了,说污点这就是个4P),作用后因为轨道数守恒一定还是四个轨道。一个轨道填的还是两个电子,叠加前每个pz里各1个电子(价键理论就会认为它们两两一对成π键了,也就无法解释离域π键了),因此形成新轨道后会填满底下的一半。
这次不让大家先考虑了,我们直接看结果。
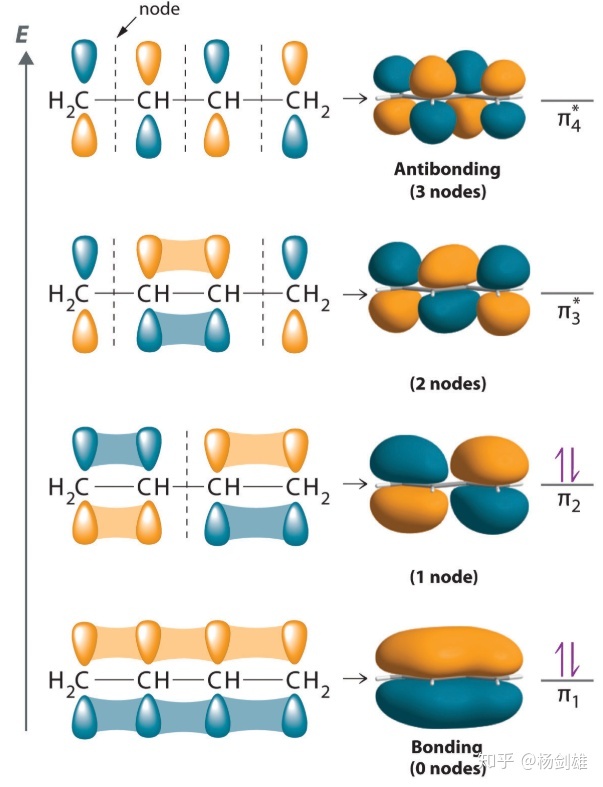
这图画得很清晰了,我们就用它来说。这里我们就能看到,在叠加前,四个pz有各种各样的组合,那么用和刚才类似的办法想想,什么情况下形成的分子轨道最稳定呢?没错,就是全都发生融合的时候。最好的情况就是四个pz都是上正下负,这样就能发生3次融合,没有节点。我把这种情况记为3 Ov 0 Nd,如果你嫌麻烦可以简写成(3,0)。
那么其次呢?那就得有且仅有一个节点,此时融合就会少一个,也就是2 Ov 1 Nd。如果不考虑对称性,其实有两种情况:四个pz的上半扇分别是(正正负负)或者(正正正负)。所以这就是叠加/节点法和对称性的不兼容之处了,知道对称性必然会排除后者,因为中间两个碳可以和两边的取消组合,后者的情况下两边是i对称(对称中心),中间两个不是。
之后就是1 Ov 2 Nd了,类似地根据对称性,必须把仅剩的融合安排在中间,两边的相和中间不同就行了。最后最不稳定的情况是互相错开,全部为节点。
所以这种偶数个碳形成离域π键的情况就完全清楚了:如果碳数为N,那么N个pz总共形成N个π分子轨道,最稳定的是(N-1) Ov 0 Nd,之后依次-1 Ov和+1 Nd,直到0 Ov (N-1) Nd为止。N个电子填满底下的一半轨道,所以HOMO是第N/2个,是N/2 Ov (N/2-1) Nd。LUMO比它高一个,Ov和Nd数反过来。
好,现在看奇数个碳的情况。这里有个问题就是因为pz上默认应该是一个电子,那奇数个碳就会出现自由基的情况,多重度变为2,我们一般不考虑。我们考虑的一般是在此基础上少一个电子或者多一个电子的情况,分别为碳正离子和碳负离子。最简单的情况是烯丙基正离子,结构就不画了,大家应该清楚。
那么在给出π分子轨道之前,大家可以先试着用类似的办法来推算一下。最稳定的情况必然是2 Ov 0 Nd,之后是1 Ov 1 Nd,最后是0 Ov 2 Nd。不过画一下图的话大家就会发现:1 Ov 1 Nd的情况对称性出现麻烦了。
这其实就是叠加/节点法和对称性匹配原则发生直接冲突的地方。怎么解决呢?我们不妨先枚举一下这三个pz线性组合的所有情况,反正就八种,也不多:(pz的上半扇)+++,++-,+-+,+--,-++,-+-,--+,---。
因为分子轨道的符号可以完全调换,所以把互相完全调换符号的情况排除一半,这样只剩+++、++-、+-+和+--。
然后再看一下这四种情况的融合和节点数,分别是(2,0)(1,1)(0,2)和(1,1)。咦,有两个1 Ov 1 Nd,难道是简并?不可能啊,总共应该就三个轨道,而且++-和+--应该也是一样的,把一个旋转一下然后完全调换符号就是另一个了。
好了,现在看一下烯丙基正离子的分子轨道吧。
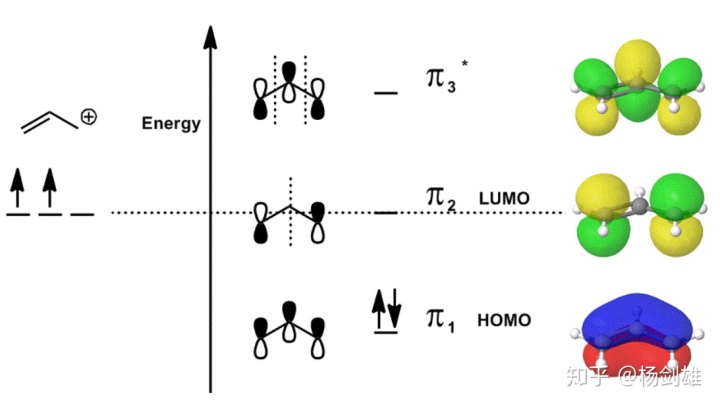
可以看到,2号轨道的中间竟然是空的!
从对称性角度其实挺好理解,那么从叠加/节点法呢?这就是这个方法的另一个亮点了:把++-和+--线性叠加,因为二者发生概率相同,所以中间的+和-抵消为0,最终变成+0-。
这样单独从外表上似乎只能看出一个远距节点,看不出融合,但我们可以这样想:空白代表正负均可。试一试,不管中间你是正还是负,这个轨道始终是1 Ov 1 Nd。
这样理解的话,从此看到这种空白就再也不怵了,理解成“正负均可”就能和前面的融合和节点完美结合,并且空白的存在还完美解决了叠加/节点法和对称性匹配原则的冲突!
所以前面的“三个轨道叠加形成一个成键、一个反键和一个非键轨道”也好理解了,至于非键是HOMO还是LUMO就要看总电子数了。
不过需要强调的是,叠加/节点法只能定性判断分子轨道的形状,所以不要用此法定量地预言分子轨道!比如,有人看了我的文章可能就会认为烯丙基正离子的最稳定的轨道中三个p轨道的贡献完全一致(因为在线性组合时p轨道的大小的确一致),但其实不是的,因为2号轨道的空白,为了满足所有pz的贡献总和一致,中间的系数肯定不能和两边完全一致。这个就需要定量计算了。
(此外顺便提一句,乙硼烷的每个3c-2e键也可以用类似的办法理解,虽然它不是离域π键,而且B原子使用的轨道是sp3杂化轨道,但原理是相通的,不信去查它的三个分子轨道。)
那么接下来我们就实战一下,定性地预测五个碳形成离域π键时的情况,还是碳正离子。
思考时间剩余 10
思考时间剩余 9
思考时间剩余 8
思考时间剩余 7
思考时间剩余 6
思考时间剩余 5
思考时间剩余 4
思考时间剩余 3
思考时间剩余 2
思考时间剩余 1
公布答案。五个轨道叠加前的p轨道上半扇分别为+++++(4 Ov 0 Nd),++0--(3 Ov 1 Nd),+0-0+(2 Ov 2 Nd),+-0+-(1 Ov 3 Nd)和+-+-+(0 Ov 4 Nd)。到这里大家应该会发现规律了:越往中间0会越多,最好的办法就是多写几种可能的情况然后找共同点,并且还得有一定的对称性。
看一下实际情况,完全一致。3号轨道的两个0的确是怎么安排正负都是2 Ov 2 Nd。
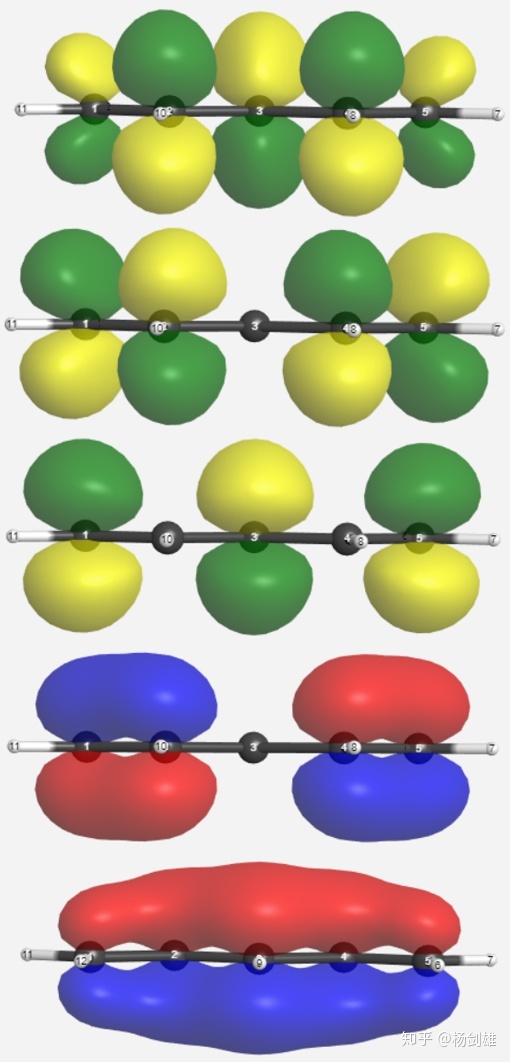
从图里看到实际分子轨道中不互相处于对称位的碳在每个分子轨道中的贡献都不一样。
所以到现在,大家应该熟悉这个+0-的套路了,因为所有的0都是在不同的符号中间——这其实很好理解,正负之间的0不管取什么都是1融合1节点,但要是+0+的话那就是天壤之别了。
在进行下一个话题之前我们先来看一个实际应用:就是刚刚提到的周环反应。
周环反应里有一类H[1,j] σ迁移反应,机理如下:
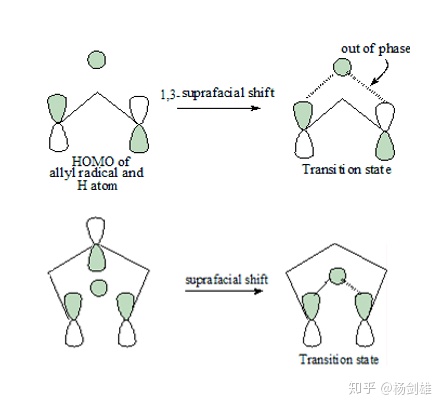
这两个例子中,我们可以看到1,3和1,5迁移在基态时分别是异面迁移和同面迁移,这一差异的本质就是两端p轨道的相位情况,相位相同就是同面迁移。这里的过渡态就是奇数个C原子的离域π键,因为两端p轨道的其中一个其实曾经是正常的C-H键,所以有一对电子,那么如果有N个碳,那电子数就是N+1,HOMO是第(N+1)/2个轨道。
先看一下1,3-σ迁移,这里HOMO就是2号轨道了,是那个+0-。所以两端相位不同,是异面迁移。之后1,5-σ迁移中,HOMO是3号轨道,上面的例题答案里有图,是+0-0+。发现了吧,第(N+1)/2个轨道作为HOMO都是(N-1)/2个融合和节点,由于+0-结构正好带有一个融合和一个节点,那就直接+0-堆起来就完事了。但是,如果两个+0-堆起来的话,结构应该是+0-0+,两端p轨道相位相同,所以1,5-σ迁移是同面迁移,但要是再堆一个+0-,也就是1,7-σ迁移,就回到异面了。所以,偶数个+0-结构末端p轨道相位相同,是同面迁移。因为+0-结构的个数是(N-1)/2,那么(N-1)/4是整数时就是同面迁移,整理一下就是:1,(4n+1)-σ迁移是同面迁移,1,(4n-1)-σ迁移是异面迁移。这样就算冷不丁让你画一个1,25-σ迁移的轨道图你都能信手拈来了,虽然这在实际上基本是不可能的。
5 环状离域π键的分析
接下来就尝试着用叠加/节点法解释环状离域π键吧。嗯,第一个例子不用说大家都知道是苯。
先用刚才的Ov Nd办法想一想。这次是环状结构了,所以1号轨道是6 Ov 0 Nd了。之后是2号轨道……大家发现问题了吧,环状结构不可能只有一个节点。道理很简单,一个节点两侧的两个符号不同的相位无法闭合,必然会形成另一个节点。所以刚才的“之后依次-1 Ov和+1 Nd”在环状体系就应该是-2和+2了。这样2号轨道必然是4 Ov 2 Nd,那3号就是2 Ov 4 Nd,4号0 Ov 6 Nd……咦等会,还有两个呢!
但不管怎么说能量肯定就这四种,所以只有一种可能性:发生简并了。
这时候动手画图吧。1号轨道肯定就一种情况:全正。4号同理,所以用排除法只能是2和3出问题了。先从2号开始,还是先试着枚举,暂时不要想+0-的套路。
你会发现两种总体情况:+++---和++++--。合并成+++0--?不太对,看起来不对称。
现在用+0-的套路看看吧。
你会发现+++---无法插0,因为中间0的位置变号后似乎就失去了一些对称性……+++---在中间有一个对称面,这是不能轻易破坏的。
而另外一种情况也有两种插0可能,我就只画俯视图了:
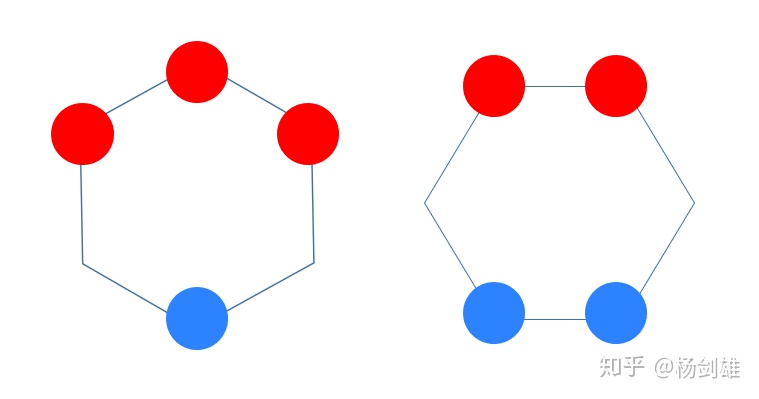
怎么看都是右边更对称一些。所以搞定了,实际情况也是一样:
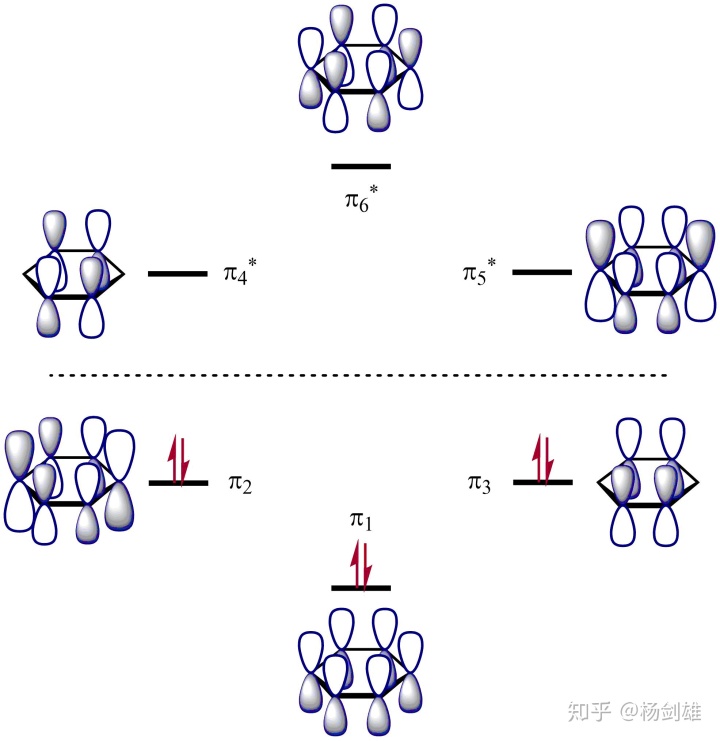
所以这次发现很难去预言分子轨道了吧,但反过来看着上图来理解就容易多了。这其实就是叠加/节点法的初衷:帮助大家从融合和节点数量的角度搞懂为什么是这个配置——如果只学了初级分子轨道理论(只有双原子分子那些)看着苯的π分子轨道肯定就像看天书一样,但知道叠加/节点法就真的一目了然。如果融合和节点数量一样,那能量就是相同的,就会发生简并。
不过到这里肯定会有人问:那你用这个方法去画环辛四烯试试?
对不起,我说了此法需要已知分子轨道,如果想尝试凭空去画的话至少也得知道分子的立体结构。环辛四烯没有芳香性,所以不是平面的,所以根本就不存在八个pz形成离域π键的情况。但如果你一定要假设它存在然后看看能不能证伪呢?那就画,具体图我就不给了,最后你会发现能量有五种,所以有三组简并,填电子后会发现HOMO上是两个未成对电子,和氧气情况相同。这里“平面环辛四烯”的HOMO其实算是非键轨道,因为在中间嘛,所以能量和pz一样。所以这样降低的能量不够多,无法补偿环张力的损失,因此环辛四烯不是平面环。其实这样解释有点牵强,但也在理,不过现在都9102年了,没有什么是计算一下解决不了的,如果不行,那就计算两下。
再来一个例子:茂。
茂其实就是环戊二烯负离子,因为是有芳香性“艹”的环“戊”二烯所以叫“茂”。汉字真的是博大精深啊,我高中学化学竞赛时第一次看到这里时简直惊呆了。好了言归正传,用类似方法可以搞出茂的π分子轨道:5 Ov 0 Nd,3 Ov 2 Nd,但接下来是1 Ov 4 Nd,没了。画不出0 Ov 5 Nd的,而且现在的样子依然遵守能量升高一级就-2 Ov +2 Nd的。然后2、3简并,4、5简并:
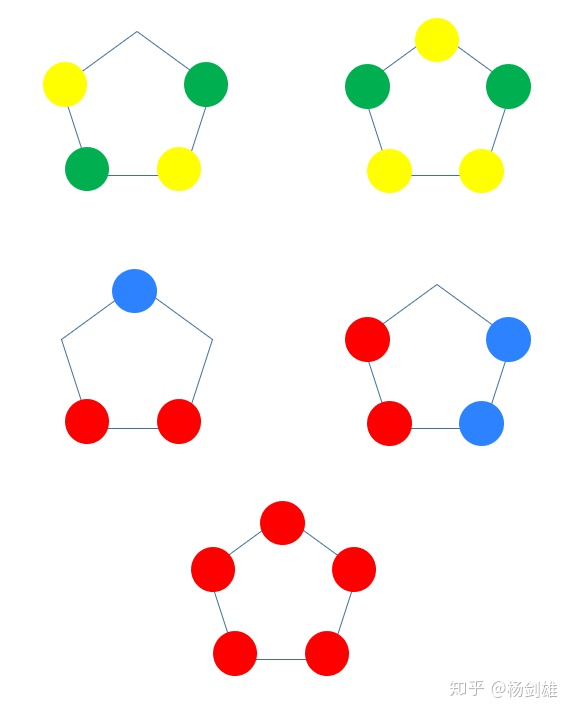
实际情况也一致。就不给图了。
不过需要说一下吡咯,它和茂是等电子体,但因为有杂原子所以失去了C5轴。这样吡咯的分子轨道就没有了简并,所以刚刚说的“如果融合和节点数量一样,那能量就是相同的,就会发生简并”是仅限于所有融合和节点发生在相同长度的键上的情况的。
结语
这样,这个神奇的叠加/节点法就被我总结完了,我确定我不是第一个提出这个方法的人,相信稍微动脑筋思考的化学老师们都会想到,但我把它推广到了用来解释一些简单的σ系统分子轨道和离域π键的π分子轨道,在后者中用融合和节点的数目判定能量高低,还解释了一些结构的π分子轨道中一些原子没有贡献的情况,引出了+0-系统,并且成功用它解释了一些问题(比如周环反应的H[1,j] σ迁移反应)。还要强调一遍,这个方法最适合用来理解已知的分子轨道,如果想用它预言分子轨道,就要时刻注意对称性的制约。
由于是第一次写化学类科普文章,各种错误和遗漏在所难免,还请广大读者批评指正。
【更新日志】
2019.08.19:首发。


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









