







在之前的文章中,已经为大家分享了几个R语言的教程,今天再为大家分享R语言的seurat包的学习笔记。
一.数据导入
本文的范例数据为seurat官网的pbmc-3k数据,文末有下载链接。当然也可以直接使用 基迪奥10X转录组结题报告中的表达量文件,如下图。
指定数据所在目录;
data_dir <-"C:/Users/MHY/Desktop/filtered_gene_bc_matrices/hg19"
载入seurat包;
library(Seurat)
读入pbmc数据;
pbmc.data <- Read10X(data.dir =data_dir)
查看稀疏矩阵的维度,即基因数和细胞数;
dim(pbmc.data)
[1]32738 2700
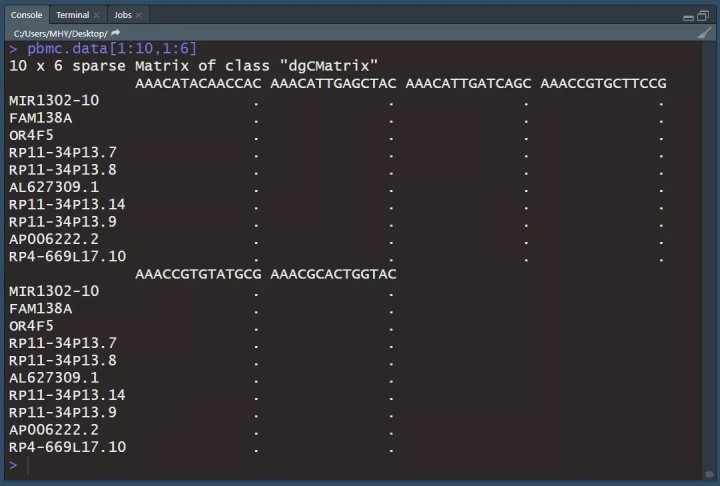
预览稀疏矩阵(1~10行,1~6列),. 表示0;
pbmc.data[1:10,1:6]
二.创建Seurat对象与数据过滤
在使用CreateSeuratObject()创建对象的同时,过滤数据质量差的细胞。保留在>=3个细胞中表达的基因;保留能检测到>=200个基因的细胞。
pbmc <- CreateSeuratObject(counts =pbmc.data, project = "pbmc2700", min.cells = 3, min.features = 200)
计算每个细胞的线粒体基因转录本数的百分比(%),使用[[ ]] 操作符存放到metadata中;
pbmc[["percent.mt"]] <-PercentageFeatureSet(pbmc, pattern = "^MT-")
过滤细胞:保留gene数大于200小于2500的细胞;目的是去掉空GEMs和1个GEMs包含2个以上细胞的数据;而保留线粒体基因的转录本数低于5%的细胞,为了过滤掉死细胞等低质量的细胞数据。
pbmc <- subset(pbmc, subset =nFeature_R
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章















 972
972











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









