






Bulk RNA-seq 分析的一个重要任务是分析差异表达基因,我们可以用 omicverse包
来完成这个任务。在omicverse中,除了最简单的ttest外,在这里,我们介绍一种类似R语言中的Deseq2等包的模型来计算差异表达基因。
原教程地址:https://omicverse.readthedocs.io/en/latest/Tutorials-bulk/t_deseq2/
环境的下载
在这里我们只需要安装omicverse环境即可,有两个方法:
- 一个是使用conda:
conda install omicverse -c conda-forge - 另一个是使用pip:
pip install omicverse -i https://pypi.tuna.tsinghua.edu.cn/simple/。-i的意思是指定清华镜像源,在国内可能会下载地快一些。
详细的安装教程见omicverse官网。
导入包
我们首先导入分析需要用到的所有包,包括omicverse, pandas, numpy, scanpy matplotlib 和 seaborn.
import omicverse as ov
import pandas as pd
import numpy as np
import scanpy as sc
import matplotlib.pyplot as plt
import seaborn as sns
#设定绘图格式,分辨率300dpi等
ov.utils.ov_plot_set()
下载基因集
当我们需要转换基因 id 时,我们需要准备一个映射对文件。在这里,我们预处理了6个基因组 gtf 文件和生成的映射对,包括 T2T-CHM13,GRCh38,GRCh37,GRCm39,danRer7和 danRer11。如果需要转换其他 id,可以使用 gtf 将文件放在 genesets 目录中生成自己的映射。
ov.utils.download_geneid_annotation_pair()
读取数据
data=pd.read_csv('https://raw.githubusercontent.com/Starlitnightly/ov/master/sample/counts.txt',index_col=0,sep='\t',header=1)
#replace the columns `.bam` to ``
data.columns=[i.split('/')[-1].replace('.bam','') for i in data.columns]
data.head()
值得注意的是,我们的数据集并没有经过任何处理,featurecounts比对时用的gtf为GRCm39,所以我们这里用GRCm39来做基因id映射
基因id转换
data=ov.bulk.Matrix_ID_mapping(data,'genesets/pair_GRCm39.tsv')
data.head()
差异表达分析
我们可以非常简单地通过omicverse进行差异表达基因分析,只需要提供一个表达式矩阵。我们首先创建一个 pyDEG 对象,并使用drop_duplicates_index去除重复的基因。由于部分基因名相同,我们的去除保留了表达量最大的基因名。
dds=ov.bulk.pyDEG(data)
dds.drop_duplicates_index()
print('... drop_duplicates_index success')
现在我们可以从表达矩阵中计算差异表达基因,在计算前我们需要输入实验组和对照组。在这里,我们指定 4-3和4-4为实验组,1--1, 1--2为对照组,我们设定method为DEseq2也是支持的,不过流程可能会有一些区别,我们放到下一期讲。
treatment_groups=['4-3','4-4']
control_groups=['1--1','1--2']
result=dds.deg_analysis(treatment_groups,control_groups,
method='DEseq2')
result.head()
Fitting size factors...
... done in 0.00 seconds.
Fitting dispersions...
... done in 1.59 seconds.
Fitting dispersion trend curve...
... done in 2.82 seconds.
logres_prior=1.1538905878789707, sigma_prior=0.25
Fitting MAP dispersions...
... done in 1.57 seconds.
Fitting LFCs...
... done in 1.27 seconds.
Refitting 0 outliers.
Running Wald tests...
... done in 1.33 seconds.
Log2 fold change & Wald test p-value: condition Treatment vs Control
在计算完差异表达基因后,我们会发现一个重要的事情,就是低表达基因有很多,如果我们不对其进行过滤,会影响后续火山图的绘制,我们设定基因的平均表达量大于1作为阈值,将平均表达量低于1的基因全部过滤掉。
print(result.shape)
result=result.loc[result['log2(BaseMean)']>1]
print(result.shape)
我们还需要设置 Foldchange 的阈值,我们准备了一个名为 foldchange_set 的方法函数来完成。此函数根据 log2FC 分布自动计算适当的阈值,但您也可以手动输入阈值。该函数有三个参数:
- fc_threshold: 差异表达倍数的阈值,-1为自动计算
- pval_threshold: 差异表达基因的p-value过滤值,默认为0.05,在有些情况下可以设定为0.1,意味着统计学差异不显著。
- logp_max: p值的最大值,由于部分p值过小,甚至为0,取对数后火山图绘制较为困难,我们可以设定一个上限,高于这个上限的p值全部统一。
# -1 means automatically calculates
dds.foldchange_set(fc_threshold=-1,
pval_threshold=0.05,
logp_max=6)
差异表达的结果可视化
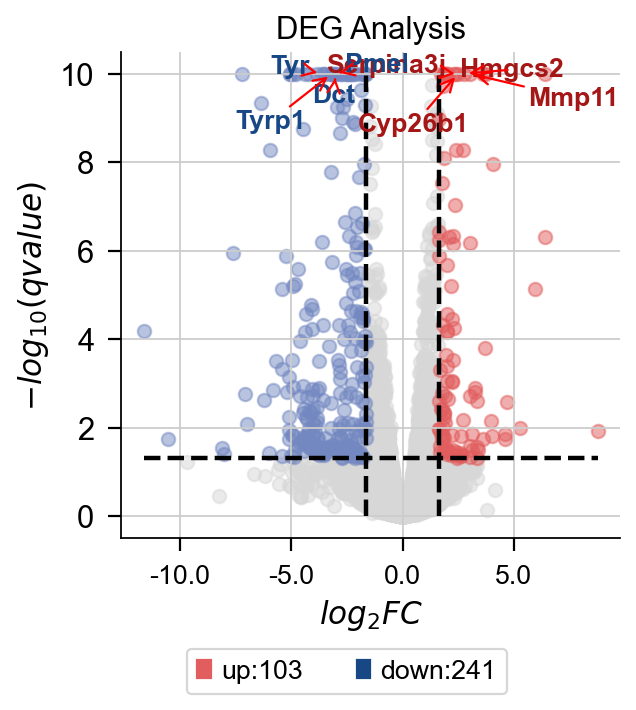
omicverse除了有较为完善的分析能力外,还有极强的可视化能力。首先是火山图,我们使用 plot_volcano函数来实现。该函数可以绘制你感兴趣的基因或高表达的基因。您需要输入一些参数:
- title: 火山图的标题
- figsize: 图像大小
- plot_genes: 需要绘制的基因,格式为list。如
['Gm8925','Snorc'] - plot_genes_num: 需要绘制的基因数,该参数与
plot_genes互斥,如果我们没有指定需要绘制的基因,可以自动绘制前n个高差异表达倍数的基因。
此外,我们还可以指定绘制的颜色等,具体的参数可以使用help(dds.plot_volcano)来查看
dds.plot_volcano(title='DEG Analysis',figsize=(4,4),
plot_genes_num=8,plot_genes_fontsize=12,)
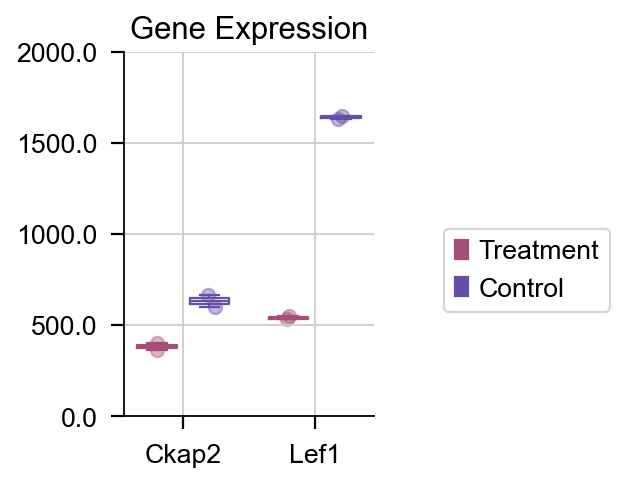
如果我们想绘制特定的基因的箱线图,我们也可以使用 plot_boxplot 函数来完成该任务。
dds.plot_boxplot(genes=['Ckap2','Lef1'],treatment_groups=treatment_groups,
control_groups=control_groups,figsize=(2,3),fontsize=12,
legend_bbox=(2,0.55))
通路富集分析
在差异表达基因计算出来后,我们需要直接进行的下一步分析往往是看差异表达的基因与哪些通路相关,这里我们常用的方法是富集分析。omicverse可以一键完成富集分析并且可视化。
我们封装了gseapy 包进入omicverse,其中包括 GSEA 富集分析的相关功能。我们优化了包的输出,并给出了一些更好看的图形绘制功能
类似地,我们首先需要下载通路数据库。我们已经准备好了五个基因集,可以使用 ov.utils.download_pathway_database()进行自动下载。除此之外,你还可以在以下网站找到你感兴趣的基因集: https://maayanlab.cloud/enrichr/#libraries
ov.utils.download_pathway_database()
#读取通路基因集,我们读取Wiki通路数据库
pathway_dict=ov.utils.geneset_prepare('genesets/WikiPathways_2019_Mouse.txt',organism='Mouse')
与此前选取差异表达的基因进行通路富集分析不同,在这里,我们使用基因的差异倍数和p值进行排序,进行GSEA分析。我们使用以下公式计算基因的顺序。
M e t r i c = − l o g 10 ( p a d j ) s i g n ( l o g 2 F C ) Metric=\frac{-log_{10}(padj)}{sign(log2FC)} Metric=sign(log2FC)−log10(padj)
rnk=dds.ranking2gsea()
们使用 ov.bulk.pyGSEA 构造一个 GSEA 对象来执行通路富集分析。
gsea_obj=ov.bulk.pyGSEA(rnk,pathway_dict)
enrich_res=gsea_obj.enrichment()
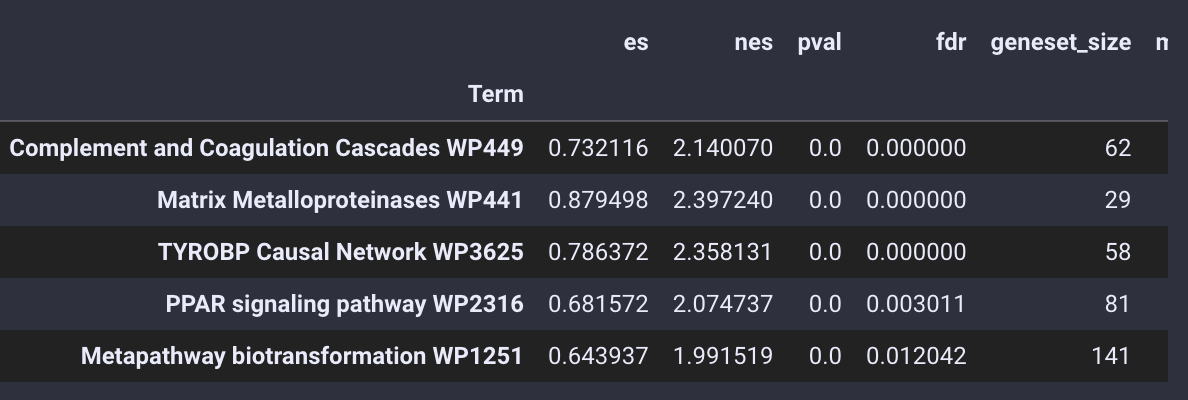
通路富集分析的结果被存放在enrich_res变量中,我们使用.head()函数来查看
gsea_obj.enrich_res.head()
为了可视化通路富集的结果,我们使用.plot_enrichment函数来完成该目的.
- num: 需要展示的通路富集结果,默认是前10条.
- node_size: 点的大小标注值. 默认是[5,10,15].
- cax_loc: 图注的横坐标. 默认值 2.
- cax_fontsize: 图注的字体大小,默认值 12.
- fig_title: 富集结果的标题.
- fig_xlabel: 富集结果的横坐标标题,默认值是’Fractions of genes’.
- figsize: 图像大小,默认值是(2,4).
- cmap: 图像颜色条,默认值是’YlGnBu’.
gsea_obj.plot_enrichment(num=10,node_size=[10,20,30],
cax_loc=2.5,cax_fontsize=12,
fig_title='Wiki Pathway Enrichment',fig_xlabel='Fractions of genes',
figsize=(2,4),cmap='YlGnBu',
text_knock=2,text_maxsize=30)

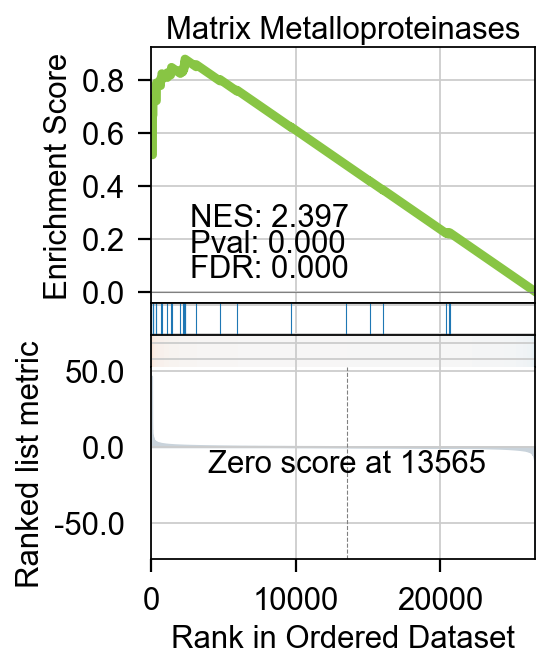
不仅仅是基本的富集分析,pyGSEA 还可以帮助我们用绘制GSEA的排序图,我们需要选择想绘制的通路,给定通路的Term位置,例如0 是Complement and Coagulation Cascades WP449 ,1 是 Matrix Metalloproteinases WP441
gsea_obj.enrich_res.index[:5]
Index(['Complement and Coagulation Cascades WP449',
'Matrix Metalloproteinases WP441', 'TYROBP Causal Network WP3625',
'PPAR signaling pathway WP2316',
'Metapathway biotransformation WP1251'],
dtype='object', name='Term')
我们可以使用.plot_gsea完成绘制。
- term_num: 需要绘制的通路在index的位置
- gene_set_title: 通路标题,可自定义,如果原index中的太长
fig=gsea_obj.plot_gsea(term_num=1,
gene_set_title='Matrix Metalloproteinases',
figsize=(3,4),
cmap='RdBu_r',
title_fontsize=14,
title_y=0.95)















 1293
1293











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









