







m6AmPred:基于序列衍生信息识别RNA N6,2′-O-二甲基腺苷(m6Am)位点
分区:2区
IF:3.864
数据:https://www.ncbi.nlm.nih.gov/geo/
网站:https://www.xjtlu.edu.cn/biologicalsciences/m6am
前言
N6,2′-O-二甲基腺苷(m6Am)是一种广泛存在于各种RNA分子上的可逆修饰。m6Am的生物学功能尚不清楚,尽管最近的研究揭示了它对细胞基因命运的影响。精确识别核糖核酸上的m6Am位点对于理解其生物学功能至关重要。我们在这里介绍m6AmPred,这是第一个用于从核糖核酸的一级序列中识别m6Am位点的网络服务器。m6AmPred建立在带有Dart算法的极端梯度增强(XgbDart)和EIIP-EIIP-PseEIIP 编码方案的基础上,当通过10倍交叉验证和独立测试数据集进行测试时,AuC大于0.954,实现了令人鼓舞的预测性能。为了严格测试和验证m6AmPred的性能,对来自两个数据源的实验验证的m6Am位点进行了交叉验证。
一、介绍
在过去的几年里,RNA的动态表观遗传修饰已经成为生物学研究的一个重要焦点。自从发现第一个结构修饰的核苷以来,已经有超过170个转录后修饰被表征。几乎所有类型的RNA都发现了RNA修饰,包括mRNA、rRNA、tRNA和snRNA,它们在调节生物功能方面表现出高度的特异性和效率。
N6,2′-O-二甲基腺苷(m6Am)是mRNA最普遍的转录后修饰之一。尽管对m6Am的生物学功能仍知之甚少,但最近的研究已经开始揭示m6Am在增强mRNA稳定性以及阻碍结直肠癌干细胞能力方面的功能。m6Am位点的准确鉴定对阐明其生物学功能至关重要,因此,一些实验方法如MeRIP-seq、miCLIP-Seq、m6ACE-seq已被开发用于鉴定m6Am位点。
然而,m6Am位点的RNA测序数据在文献中仍然非常有限,主要是因为网络实验室实验中用于免疫沉淀的抗体成本高、实验时间长、特异性低。
在这项研究中,我们试图开发一种计算机技术来识别核糖核酸序列中的m6Am位点,使用一种极端梯度增强算法,XgbDart作为分类器。
已经进行了一些尝试来开发预测核糖核酸表观遗传修饰的计算方法。然而,据我们所知,到目前为止还没有m6Am站点预测器可用。
因此,在这项研究中,我们想提出第一个m6Am预测因子m6AmPred,它可以用于精确识别RNA序列中的M6Am位点。为了便于访问我们的预测器,还开发了一个用户友好的网络服务器,并在https://www.xjtlu.edu.cn/biologicalsciences/m6am.提供。预计我们的m6Am站点预测器m6AmPred可以最好地利用有限的实验验证数据,并通过提供可靠的计算方法来帮助m6Am站点修改的研究。
二、材料与方法
2.1 训练集和测试集
阳性数据集(m6Am位点)从最近发表的单核苷酸分辨率m6Am测序数据中获得,测序结果通过miCLIP-seq技术在HEK293细胞系上产生。数据从基因表达综合数据库(GEO)下载,GEO登录号为GSE63753和GSE78040。先前的研究显示长度为41个核苷酸[nt]且中间有一个RNA修饰位点的RNA序列显示出最佳结果,因此我们最初采用了这个公式,并通过生成41nt序列来设计我们的阳性数据集,其中实验鉴定的Am位点位于中心。从阳性m6Am位点的相同转录物上的未修饰BCA基序中随机选择未修饰的Am位点(阴性数据),并产生10个阴性数据集。通过将这10个负集合中的每一个集合与正数据相结合,以1:1的正负比率构建了10个独立的数据集。在评估期间对它们的预测性能进行平均,以减少批次差异。
来自两个GEO数据集的数据被组合,然后被分离成完整的转录本数据集和成熟的RNA数据集,结果在完整的转录本数据集中有2447个阳性序列,在成熟的RNA数据集中有1673个阳性序列。完整的转录本和成熟的核糖核酸数据集被随机分成训练集和测试集,比例为8:2。然后,将两个数据集分别作为训练集和测试集,测试模型的鲁棒性。表1列出了每个数据集的位点数量。
2.2 特征提取
机器学习算法的高分类精度在很大程度上依赖于用于RNA序列特征提取的序列编码策略。
为了获得最佳性能,本研究采用了两种编码策略,即核苷酸化学性质(NCP) +核苷酸密度(ND)和电子-离子相互作用势(EIIP) +伪EIIP(pseiip),并对它们的性能进行了比较。
2.2.1.核苷酸化学性质和核苷酸密度
核苷酸密度(ND)表示核苷酸在每个位置的分布和频率信息。第N位核苷酸的密度(di)可以通过第(i + 1)位之前出现的N的数量(N)除以i :,di= n/i来计算。因此,对于序列“AUAGUCAUAA”,A的密度在第1、3、7、9和10位分别为1、0.67、0.43、0.44和0.50。
类似地,位置2、5、8处的U为0.50、0.40、0.38,位置6处的C为0.17,位置4处的G为0.25。
核苷酸化学性质的编码方案是基于四个核糖核酸核苷酸的不同化学结构而设计的。核糖核酸的四个组成部分,腺苷(A)、尿苷(U)、鸟苷(G)、胞嘧啶©根据环结构的数量(两个用于A、G,一个用于C、T)、氨基(A、C)或酮基(G、T)的存在、强(C、G)或弱(A、T)氢键的存在而分为三组。
基于该信息和核苷酸密度信息,来自序列S(长度为l)的核苷酸N可以由满足以下等式的公式Ni ={xi,yi,zi,di }(i = 1,2,3,…l)表示:
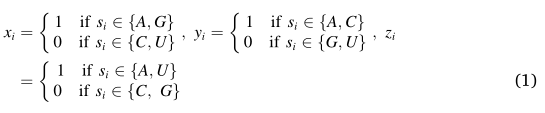
具体地,A、C、G、U可以分别被编码为向量(1,1,1,di),(0,1,0,di),(1,0,0,di)和(0,0,1,di)。因此,一个核糖核酸序列中的每个核苷酸将被编码成四个数值。
2.2.2.电子-离子相互作用势(EIIP)和伪EIIP
在EIIP,每一个基因核苷酸都被编码成一个数值,代表它的电子-离子相互作用势。表2给出了每个核苷酸的EIIP值。

在我们的研究中,EIIP编码生成一个长度为41的数字向量。此外,通过将三核苷酸的数值和其在给定序列中的频率相乘来计算伪EIIP。因此,对于任何给定的mRNA序列,PseEIIP都是一个64维向量,如下所示:
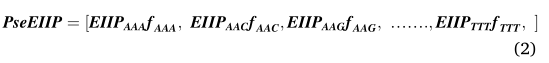
其中,fxyz为三核苷酸的频率
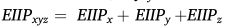
x,y,z属于{ A,G,C,T}。在我们的研究中,通过结合EIIP和PseEIIP,每个输入序列将被转换成41+64 = 105维向量。
2.3 评估矩阵
使用五个指标来评估我们的模型的性能,公式如下:

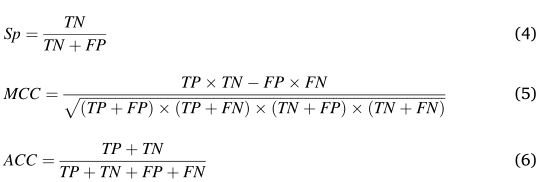
其中TP、TN、FP、FN分别代表真阳性、真阴性、假阳性、假阴性的数量。应用0.5的阈值来计算结果的敏感性和特异性。
2.4 机器学习分类器的选择
支持向量机(SVM)、随机森林、线性模型(GLM)是RNA修饰预测中最流行的机器学习分类器,已被广泛用于不同的修饰预测。
此外,极端梯度增强算法XgbDart (XGBDART)在本研究中进行了测试,该算法以前没有被其他RNA修饰预测器使用过。XgbDart最初是由K. V. Rashmi和Ran Gilad-Bachrach提出的,他们通过对系综树使用dropouts来克服过度专门化的问题。
我们通过10倍交叉验证和独立测试集评估了这些算法的性能。
2.5 模型优化
通过使用不同的输入序列长度和超参数调整,进一步优化了所选模型。核糖核酸序列(1)的长度设计为2N+1,中间有一个m6Am位点,两条链上有N (nt)个侧翼序列。表3列出了不同长度、不同氮含量的基准核糖核酸序列。

XgbDart算法的超参数通过在提升迭代次数、最大树深度、收缩率(学习率)、最小损失减少、子样本百分比、子样本列比、树丢弃的比例、跳过丢弃的概率等等来调整。选择最佳序列长度和超参数进行m6AmPred的最终构建。
2.6 特征重要性
了解机器学习预测背后的原因可以帮助评估机器学习算法,并增加人类对模型结果的信任。因此,我们分析了特征的重要性,并将结果与成熟RNA数据集中识别的基序进行了比较。成熟RNA序列的基序被DREME基序发现工具捕获。
2.7 估计m6Am RNA修饰的概率
在m6Am网络服务器中,如果一个位点的预测值高于0.5,则该站点被预测为假定的m6Am站点。然后计算m6Am位点的似然比(LR)来估计m6Am RNA甲基化的概率,最小LR值为1。LR值较大的位点表明它更有可能是m6Am位点。

三、结果
3.1 确定m6Am位点预测的最佳机器学习算法和特征提取方法
四种分类器结合不同特征提取方法的性能评估结果如图1所示。

图1。独立测试集对四个具有两种不同编码方案的分类器的性能评估。图1比较了不同编码方案的不同分类器的AUC值。左图比较了四个分类器在全转录模式下两种不同编码方案的预测性能,右图显示了成熟RNA模式下的结果。* XGBDART = XgbDart,SVM =支持向量机,RF=随机森林,GLM =线性模型。
对于分类器比较,XgbDart在所有四种算法中取得了最好的整体结果。因此,我们选择了XgbDart结合EIIP-pseiip编码策略来构建机器学习模型。
3.2 模型优化
通过将侧翼序列(flanking sequences)从5nt设置为60nt来优化基准数据集的长度。尽管较长的序列在AUC和准确性方面表现出稍好的性能,但考虑到侧翼序列的长度将是我们服务器的一个阈值,用户可能希望分析较短的序列,
我们选择了40nt进行进一步优化。模型的代码在我们的网站上提供,因此用户可以在本地选择不同的侧翼长度(图2)。

图2。通过AUC和ACC优化序列长度。通过在m6Am位点周围设置不同的侧翼序列,优化了基准数据集的序列长度。模型的性能通过AUC和ACC值进行比较。
通过网格搜索优化了XgbDart模型,超参数的最佳值如表4所示。

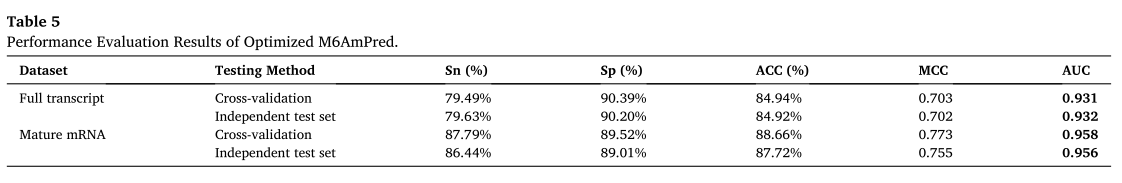
表5显示了81nt基准数据集序列和优化的XgbDart超参数的最终性能结果。
3.3 两个独立数据集的性能评估
通过使用来自两个不同来源的数据,进一步评估了我们模型的稳健性。
分别使用GSE63753数据集和GSE78040数据集作为训练集,通过10倍交叉验证评估性能,然后将另一个数据集作为独立测试集。具体来说,当使用GSE63753进行训练时,以8:2的比例随机分离,进行10倍交叉验证,并使用整个GSE78040进行独立测试,反之亦然。性能评价结果列于表6。

该模型具有较高的AUC,证明了该模型的鲁棒性和泛化能力。
3.4 特征解释
由插入符号包生成的前10个加权特征如图3d所示。
根据模型性能评估中的重要性对特征进行排序。为了验证这些特征,我们还分析了成熟RNA数据集中的基序,检测到的前3个基序如图3a-c所示。值得注意的是,当我们生成负序列时,引入了“BCA”基序,以增加正序列和负序列的相似性,因此预测者不太可能识别序列中的“BCA”基序。当比较在阳性序列中识别的重要特征和基序时,很明显我们的M6AmPred可以通过识别像CGG、GCG、CGC和阳性A位点(N41)周围的核苷酸这样的特征来捕获序列中的模式。
图3。基序成熟的核糖核酸序列和特征重要性分析。(a-c) DREME对成熟RNA序列的分析确定了三个基序:GCCAT、BCAB、GGCGGC。(M6AmPred的特征重要性分析。前10个功能按重要性顺序排列。
3.5 Web实现
已经开发了一个网络服务器,以便于访问M6AmPred。可以在https://www.xjtlu.edu.cn/biol生物科学/m6am轻松访问。它允许用户以FASTA格式提交长度超过81nt的查询RNA序列,并在中心提供腺苷供评估。网络预测器将使用我们的M6AmPred预测器预测m6Am站点的可能性。所有假定的m6Am位点将被识别为具有与m6Am位点相似的胡德比率的输出,并且预测结果可以被下载用于进一步分析(图4).

我们模型的代码在我们的网站下载部分提供,允许我们的用户在本地运行模型。
四、讨论
在本研究中,我们利用来自两个不同来源的m6Am修饰位点,并建立了一个XgbDart模型来预测m6Am修饰位点。当通过独立的测试集评估时,我们的模型获得了相当高的准确性,对于完整转录AUC为0.932,对于成熟RNA AUC为0.956。该模型还通过使用来自两个来源的数据进行了交叉评估。
值得注意的是,作为一种XgBoost算法,XgbDart在运行时默认会占用所有的CPU内核。考虑到计算成本,我们将web服务器的线程限制为4个内核。但是模型的代码在我们的网站下载部分提供,这将允许我们的用户无限制地在本地运行它。
当使用GSE63753作为训练集时,10倍交叉验证的AUC和准确性出乎意料地低于使用整个GSE78040作为独立测试集的结果。与我们的一般知识不同,该模型通常比测试数据集更适合训练数据集。我们认为有两个可能的原因。首先,GSE63753数据集的质量可能较差,并且包含一些假阳性位点。
然而,该模型仍然可以从中学习一些模式,因此,当该模型应用于高质量数据集时,结果更有希望。另一种可能的解释是,GSE63753数据集在正样本和负样本之间有一些特定的相似数据点,机器学习算法很难将其分离。这种现象在之前的Iris数据集中被检测到。当有更多测序数据可用时,我们将不断提高模型的稳健性。
本研究仅考虑序列特征。需要进行进一步的研究,使用二级结构、基因组信息、核糖核酸类型等其他特征来优化模型,以进一步提高模型的鲁棒性和泛化能力。















 785
785











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









