






 本文介绍了一种新型深度学习模型APPM,专用于预测人类白细胞抗原(HLAI)I类等位基因的抗原呈现。APPM在大规模质谱数据集上训练,显著提高了预测的阳性预测值(PPV),并通过结合传统方法进一步增强准确性。研究还展示了APPM在识别驱动突变新抗原中的潜力,为个性化免疫疗法提供关键数据。
本文介绍了一种新型深度学习模型APPM,专用于预测人类白细胞抗原(HLAI)I类等位基因的抗原呈现。APPM在大规模质谱数据集上训练,显著提高了预测的阳性预测值(PPV),并通过结合传统方法进一步增强准确性。研究还展示了APPM在识别驱动突变新抗原中的潜力,为个性化免疫疗法提供关键数据。
摘要
目前预测新抗原的算法受到体外结合亲和力数据和算法的限制,不可避免地导致高假阳性。在这项研究中,我们提出了一个名为APPM(抗原呈现预测模型)的深度卷积神经网络来预测人类白细胞抗原(HLA)I类等位基因的抗原呈现。
APPM在大型质谱(MS)HLA-肽数据集上进行了训练,并用独立的MS基准进行了评估。结果显示,APPM在阳性预测值(PPV)方面优于免疫表位数据库(IEDB)推荐的方法(0.40 vs. 0.22),在结合这两种方法后,预测值将进一步提高(PPV=0.51)。我们进一步将我们的模型应用于预测来自共识驱动突变的新抗原,并确定了16000个具有 "驱动 "特征的推定新抗原。
INTRODUCE
癌症的发展是肿瘤特异性体细胞突变(1-3)的结果,其中编码区的非静止突变可以被免疫系统识别为 "外来 "的信标,被称为新抗原(4,5)。当它们通过主要组织相容性复合体(MHC)[也叫人类白细胞抗原(HLA)]呈现在癌细胞表面时,可以引起保护性的抗肿瘤反应。新抗原长期以来被认为是免疫治疗的理想目标,因为它们被肿瘤细胞限制性地表达,并且不受中央或外周耐受性的影响(6)。近年来,基于新抗原的免疫疗法取得了巨大的成功(7-11),进一步突出了准确预测新抗原对开发癌症疫苗和采用T细胞疗法的重要性(12-15)。然而,目前从突变肽中识别免疫原性新抗原的预测方法和算法还远远不能令人满意。精度低是其鉴定方案的主要障碍(16),部分原因是它们主要依赖于HLA-肽的结合亲和力(17)。通过体外实验产生的结合亲和力结合实验忽略了肽传递过程中涉及的其他生物步骤,这导致了相当一部分的假阳性.
解决这个问题的方法之一是用从单等位或混合等位癌细胞系的HLA复合物中洗脱出来的肽来训练预测算法,并通过质谱分析来确定(19)。质谱数据集描述了自然呈现在细胞表面的肽,它已经经历了抗原处理和运输的步骤(20,21)。
精度低的另一个原因可能是没有考虑到识别特征,如氨基酸特性和空间结构(22,23)。与MHCflurry、NetMHC-4.0和NetMHCpan-4.0(24-26)中使用的其他人工神经网络相比,卷积神经网络(CNN)保留了局部空间特征(27),更适用于研究氨基酸的空间位置对结合至关重要的肽(28)。
在这项研究中,我们提出了一个抗原呈递预测模型(APPM),这是一个经过训练的CNN算法,可以准确预测HLA-I分子呈递多肽的可能性。APPM在20个高频HLA等位基因中的特异性和阳性预测值方面优于IEDB推荐的方法(2020.04 netMHCpan EL 4.0)。此外,我们预测了来自TCGA驱动突变的新抗原,其制备可用于现成的免疫疗法,以节省从检测突变到个性化疫苗注射的时间。
方法
数据收集
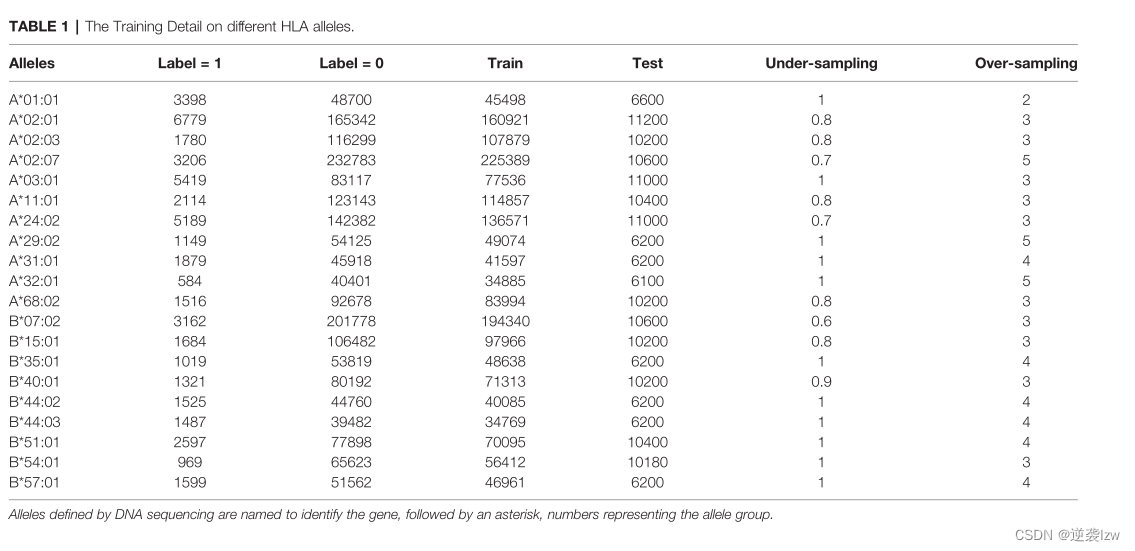
收集了超过1,900,000份已发表的单allelic或混合allelic细胞系的HLA-肽MS数据,这些细胞系共同表达了20种高频的HLA-A和HLA-B异型(16,19,29,30)。所有这些数据都是以二进制符号标注的。标签=1表示MS识别的肽(命中率),而标签=0表示参考蛋白质组(SwissProt)中没有通过质谱检测的肽。
数据编码
训练数据集是长度为8-mer到11mer的肽,由一个字母的氨基酸字母表表示(总共20个不同的氨基酸,即'ACDEFGHIKLMN PQRSTVWY')。这样的长度范围抓住了所有HLA I类限制性肽的~95%。为了实现机器学习,多肽序列通过一个单次编码方案被矢量化。具有多种长度的肽(8-mer到11-mer)通过使用填充字符'Z'被表示为固定长度的向量。每个氨基酸和填充的'Z'都被编码为一个单点矢量(详见图S1)。因此,肽被编码为11行(最大长度)由21列(20个不同的氨基酸字母和填充字符'Z')组成的固定矩阵。
训练数据集的不平衡分布
MS数据集的集合显示了严重的类别不平衡。
总的来说,0标签的数据总数为1,866,484个,是1标签的对应数据的39倍。一个极端的例子是HLA-A*02:07数据集,其中负标签的记录比1标签的记录多72倍。这种极端的不平衡影响了机器学习模型的预测,倾向于在0标记的肽上表现得更好(大多数),在1标记的肽上表现得更差(少数)(31)。因此,在预处理训练数据集时,通过过度取样和不足取样程序来调整类的平衡。简而言之,欠采样是通过随机删除0标签的训练数据点,而过度采样则是重复1标签的数据点。
表1显示了不同HLA等位基因上过度采样和欠采样的比例。
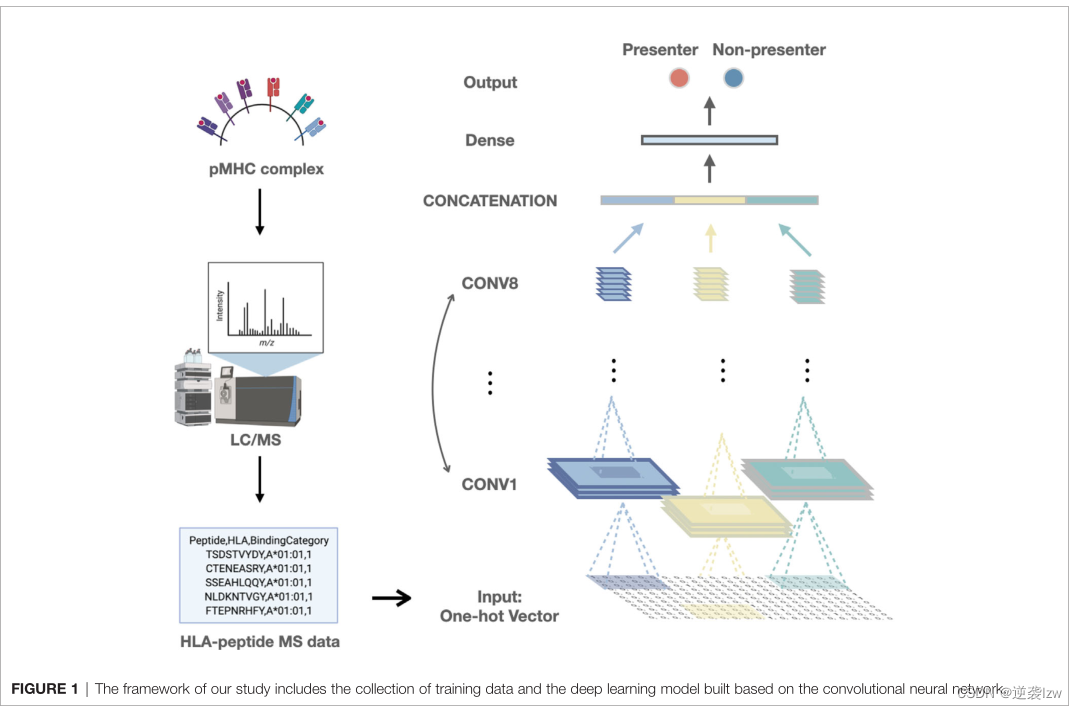
卷积神经网络
在这项研究中,使用了一个先进的CNN,其灵感来自GoogLeNet的inception模块(32, 33)。构建了三个平行的卷积段,每个卷积段有八个二维卷积核,以最大限度地提取特征(详见图S2)。三个卷积层的输出连接到一个扁平化的矩阵,并被输送到包含100个隐藏节点的全连接层。输出层显示两个节点的二元分类结果,其中测试的肽被分类为与HLA结合或不结合。
该模型用Tensorflow(1.14.0版)实现,用标准参数的Adam优化算法训练。取代经常使用的激活函数Rectified Linear Unit(ReLU),应用Leaky ReLU(a=0.2)的提前函数来激活模型,并引入 "drop-out "和 "early stopping "方案来避免过度拟合。
独立验证数据集
为了对APPM和其他HLA-肽预测器进行基准测试,我们从其他使用细胞系表达单一HLA等位基因的研究中收集了HLA结合肽的MS数据集(34,35)。从这些MS识别的肽(命中率)中,我们通过Uniprot人类参考蛋白质组(UP000005640_9606)对相同蛋白质的未观察到的肽进行采样,产生非结合物(诱饵集),如前所述(36)。对于每个MS识别的肽,我们随机选择99次诱饵肽,四个不同的长度(8,9,10,11),每个长度的数量是相同的。99次偏移的理由是,对于一个生物体的肽片段样本,通常认为大约有1%~2%的片段会与MHC受体结合(37)。在去除模型训练数据中出现的多肽和从不同的蛋白质中重复取样,我们得到了一个单等份的基准数据集。
预测性能指标的计算
敏感性,也叫召回率,计算方法是:
特异性的计算方法是
阳性预测值,也称为精确度,计算公式为
癌症基因组图谱(TCGA)驱动突变
基于测序和结构分析,我们最终选择了3437个癌症驱动基因突变作为共识名单,这些突变由CTAT-群体、CTAT-癌症或结构聚类的≥2种方法确定
来自驱动突变的候选肽
对于每个驱动突变,我们提取8-11mer的候选肽,这些肽含有驱动突变的特定氨基酸,用于新抗原筛选。例如,9-mer候选肽的提取过程描述如下(图S3)。首先,我们从蛋白质序列中提取了一个17-mer的肽,其中突变体氨基酸被置于中心位置,8个上游和下游的野生氨基酸作为侧翼。其次,通过使用滑动窗口协议,将一个9个氨基酸大小的窗口滑动N次(N = 9),以获得9-mer肽。简而言之,突变体氨基酸作为第一个9-mer肽的终点。这个9mer的滑动窗口沿着17-mer的片段移动,直到突变点成为9-mer的起点。其他长度的肽也以同样的方式处理。
结果:
发展APPM
我们旨在通过一个新的工具来提高HLA肽预测方法的精确性和特异性,该工具是在改进的训练数据和新的监督机器学习模型上训练出来的。MS数据的HLA-肽是通过免疫沉淀HLA分子洗脱出来的,然后通过液相色谱-串联质谱(LC-MS/MS)鉴定(40,41)。与体外结合亲和力试验相比,MS数据直接描述了细胞或组织主动呈现的肽(42)。我们从16个单等位HLA-A和HLA-B细胞系中收集了公开的HLA-肽的MS数据,这些细胞系是通过遗传工程来表达单一的HLA等位基因,以及从表达多个HLA复合等位基因的B淋巴细胞或癌症细胞系中收集的(16, 19, 29, 30, 43)。这些MS数据包括20个高频的HLA-I等位基因。我们将数据集分成三组:训练、验证和测试组(方法)。由于有如此多的阴性肽(来自参考蛋白质组),我们采用了过度采样和不足采样的方案,这样可以中和相当一部分的不平衡问题。利用这些公开的HLA-肽MS数据,我们建立了一个卷积神经网络(CNN)框架来预测HLA-I的呈现,这是一种深度学习的形式,擅长处理一般的序列数据,如氨基酸序列(图1)(28)。该模型有三个平行的卷积模块,每个模块由八个二维卷积层组成,保留了HLA I类-肽结合的特征。
模型表现:
我们得到NetMHCpan4 EL的平均特异性和阳性预测值(PPV)为0.97和0.22(补充文件1)。当对相同的数据进行测试时,APPM的特异性为0.99,PPV为0.40,超过了NetMHCpan4 EL的表现。在减少假阳性率方面的改进是巨大的,PPV平均增加了80%对于HLA I类的20种常见单倍型,APPM只在HLAA*02:01上表现出比NetMHCpan4 EL略低的PPV,但在其余19种HLA单倍型上表现出更高的PPV,尤其是HLA-A*02:03、HLA-A*29:02、HLA-A*32:01和HLAB*40:01,增加了1倍以上(图2B),表明我们算法的优势。
结合算法可提高预测性能
有趣的是,在APPM和NetMHCpan4 EL之间观察到假阳性肽的低重叠率(19%)(图3A),可能是由于不同的预测机制。在这种情况下,我们假设,通过结合这两种预测方法,可以提高预测性能。我们重新定义了预测结果:只有在两种方法中都确定为阳性的肽才被视为阳性。使用联合预测,我们得到的PPV为0.51(图3B),明显高于APPM和NetMHCpan4 EL(图3C,p = 0.013,t检验和图3D,p < 0.001,t检验),而灵敏度没有明显下降(图3E,p = 0.1,方差分析)。这些结果表明,不同算法的综合预测可以提高新抗原选择的阳性率,这与以前的研究一致(45,46)。
特定等位基因的表现形式
为了说明HLA-I等位基因与肽的结合特点,我们为20个HLAI等位基因绘制了等位基因特异性表达图案(所有等位基因的图案见补充文件2)。
与以前的研究一致(17,19,47),这些图案揭示了HLA呈现对8-11长度的多肽的每个序列位置的依赖性(图4A)。例如,9mer的锚定残基是第2位的氨基酸(指P2,其他位置的类似缩写)和P9,而11mer在P2和P11
与以前的工作相比(48),一些不同的HLA等位基因有类似的呈现图案。例如,HLA-A*02:01和HLA-A*02:03具有相同的结合特异性,即口袋优先结合在P2位置的亮氨酸和最后位置的缬氨酸/亮氨酸的肽。同样,HLA-A*03:01和HLA-A*11:01在最后一个位置呈现赖氨酸,而HLA-B*40:01、HLA-B*44:02和HLA-B*44:03更喜欢传递P2位置有谷氨酸的肽(图4B)。
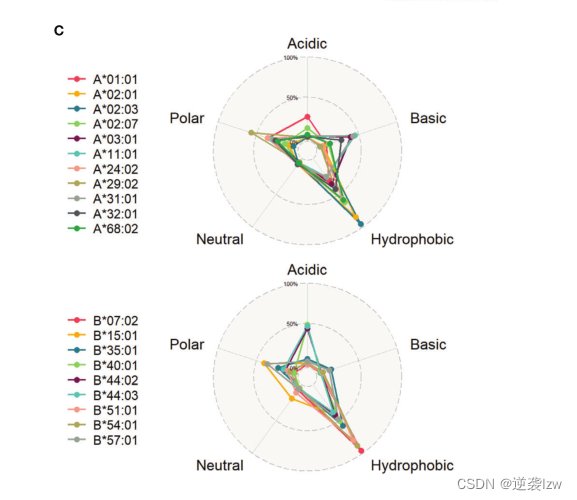
此外,我们分析了20个HLA等位基因的锚定残基的氨基酸特性,并完善了它们的结合特性:这些结合肽在锚定残基处富含疏水氨基酸。这与已知的HLA-I结合和呈现的偏好是一致的(23,49)。我们还探讨了HLA-A和HLA-B分子之间对锚定残基的氨基酸特性的整体偏好(图4C)。除了对疏水性氨基酸的共同偏好外,HLA-A等位基因更喜欢结合碱性和极性氨基酸,而HLA-B等位基因更喜欢酸性氨基酸。
讨论
在这项研究中,我们通过在体外MS数据和CNN深度学习模型上训练模型来建立高PPV新抗原预测算法。基于单等位基因的基准,我们证明我们的模型APPM在19个高频HLA等位基因中的精度优于netMHCpan4 EL。此外,APPM和NetMHCpan4 EL的组合提高了预测性能,这表明组合策略可以更精确地识别临床实践中的潜在新抗原。然而,质谱分析本身有一个技术上的局限性:不是所有可能的洗脱配体都能被检测到,这不可避免地产生了假阴性肽(53-55)。
这项工作的一个重要限制是,我们应用MS数据集来训练和评估我们的预测器。使用MS识别的多肽来反映基因表达、蛋白酶裂解、运输和呈现的因素可能会给我们的预测带来MS偏差。我们的工作也忽略了T细胞对呈现的表位的识别。我们的预测器所确定的许多推定的新抗原在用于癌症患者时不会诱发CD8+T细胞反应。这一局限性与之前的研究一致,即抗原的呈现对于诱导强大的抗肿瘤反应是必要的,但不是充分的(56)。
数据和代码网址:
https://github.com/haoqing12/APPM.git
补充材料:
这篇文章的补充材料可以在网上找到:https://www.frontiersin.org/articles/10.3389/fimmu.2021.
682103/full


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









