






之前我们对pathview包有了一个初步的了解,并学习了如何快速上手这个包,今天我们来进一步学习这个包的功能。
1.主函数pathveiw()
pathview()函数是包中最重要的函数,因此为了更熟练使用这个包,我们应该对这个函数做一个全面的了解。
该函数的主要参数如下:
pathview(gene.data = NULL,
cpd.data = NULL,
pathway.id,
species = "hsa",
kegg.dir = ".",
cpd.idtype = "kegg",
gene.idtype = "entrez",
gene.annotpkg = NULL,
min.nnodes = 3,
kegg.native = TRUE,
map.null = TRUE,
expand.node = FALSE,
split.group = FALSE,
map.symbol = TRUE,
map.cpdname = TRUE,
node.sum = "sum",
discrete=list(gene=FALSE, cpd=FALSE),
limit = list(gene = 1, cpd = 1),
bins = list(gene = 10, cpd = 10),
both.dirs = list(gene = T, cpd = T),
trans.fun = list(gene = NULL, cpd = NULL),
low = list(gene = "green", cpd = "blue"),
mid = list(gene = "gray", cpd = "gray"),
high = list(gene = "red", cpd = "yellow"),
na.col = "transparent",
...)我也对所有参数的含义做了简要注释,表格如下。大家可以快速浏览一遍,有个印象。在实际使用过程中或是看下面的讲解时对某些参数有疑惑,可以返回来看看。
参数 | 意义或用途 |
gene.data = NULL, | 输入的时数据。 向量(单样本)或类似矩阵的数据(多样本)。向量应该是数字,以基因ID作为命名,也可以是基因ID的字符。字符向量被视为离散或计数数据。 |
cpd.data = NULL, | 化合物的数据,与gene.data类似。ID要与KEGG compound ID相对应。 |
pathway.id, | 要展示的或要映射的通路,通常使用5个字符的KEGG ID |
species = "hsa", | 物种信息,默认为人类 |
kegg.dir = ".", | 结果图保存的地址,默认为当前的工作路径 |
cpd.idtype = "kegg", | 化合物数据中使用的ID类型,默认为标准的KEGG化合物ID,包括化合物、聚糖以及药物的ID |
gene.idtype = "entrez", | 基因数据中使用的基因ID类型,默认为标准的KEGG基因ID |
gene.annotpkg = NULL, | 其他的基因注释包,用于ID转换,一般情况下不使用 |
min.nnodes = 3, | 通路图最小的节点数 |
kegg.native = TRUE, | 当为TRUE时,结果图是KEGG原始图,png格式;当为FALSE时,结果图是由Graphviz引擎绘制,pdf格式。 |
map.null = TRUE, | 是否要在通路图上绘制NULL数据,当 kegg.native = TRUE才会产生影响 |
expand.node = FALSE, | 在 kegg.native = FALSE时,是否将多个基因节点变为一个基因节点,此节点会继承其它节点的所有关系 |
split.group = FALSE, | 在 kegg.native = FALSE时,是否将属于同一基因节点的基因分为数个节点 |
map.symbol = TRUE, | 在 kegg.native = FALSE时,在结果图中是否将ID转换为Symbol。在 kegg.native = TRUE时,默认使用KEGG ID |
map.cpdname = TRUE, | 在 kegg.native = FALSE时,在结果图中是否将ID转换为标签。在 kegg.native = TRUE时,默认使用KEGG ID |
node.sum = "sum", | 当多个基因或化合物对应同一个节点时,用什么方法处理数据。默认为求和。其他还有平均值或中位数等 |
discrete=list(gene=FALSE, cpd=FALSE), | 一个包含两个逻辑值的列表;取决于基因或化合物中的数据是连续性变量还是分离型变量 |
limit = list(gene = 1, cpd = 1), | 一个列表;决定基因和化合物数据在图像中的图例数值范围 |
bins = list(gene = 10, cpd = 10), | 一个列表;决定基因和化合物数据图例分成几度 |
both.dirs = list(gene = T, cpd = T), | 一个包含两个逻辑值的列表;取决于数据是单方向性(如0~1)的还是双方向性的(如-1~1) |
trans.fun = list(gene = list(gene = "gray", cpd = "gray"), | 一个列表;决定如何处理基因和化合物数据,可以用log、abs函数,或其它自定义函数 |
low = list(gene = "green", cpd = "blue"), | 一个列表;决定了基因数据和化合物数据图例中最低值的颜色 |
mid = list(gene = "gray", cpd = "gray"), | 一个列表;决定了基因数据和化合物数据图例中中间值的颜色 |
high = list(gene = "red", cpd = "yellow"), | 一个列表;决定了基因数据和化合物数据图例中最高值的颜色 |
na.col = "transparent", | NA数据的填充颜色 |
2.输出形式
Pathview在通路图数据的可视化时有两种输出格式,分别是原始的KEGG视图和Graphviz视图。
前者将用户的数据呈现在原始的KEGG路径图上,因此很自然,更容易阅读,使用的也是默认的KEGG ID,输出的图像时PNG格式。
后者使用Graphviz引擎布置路径图;因此可以更好地控制节点或边的属性和路径拓扑结构,输出的图像时PDF格式。在使用时可以根据实际需要选择合适的输出格式。
此外Pathview的绘图可以调整图层,来加速图像的生成。下面我们分别学习一下使用方法:
2.1two-layer graph,两个图层绘图
在之前的例子中,我们绘制的图像只有一个图层,节点颜色在原始KEGG图上被修改,而原始的KEGG节点标签(节点名称)则保持不变,即显示的是基因的KEGG ID。这样能使输出的图像最小,但是相对的计算时间就会变长。
如果我们不在意大小,只想快点看到结果,可以设置same.layer = FALSE参数,这样相应的节点颜色和标签会添加在另一个图层当中,速度有很大提升。此时图像上的的KEGG基因标签会变成gene symbols,也更符合我们的平时的习惯。
pv.out <- pathview(gene.data = gse16873.d[, 1], pathway.id = demo.paths$sel.paths[i],
species = "hsa", out.suffix = "gse16873.2layer", kegg.native = T,
same.layer = F)在我们设置same.layer=FALSE后,结果如下: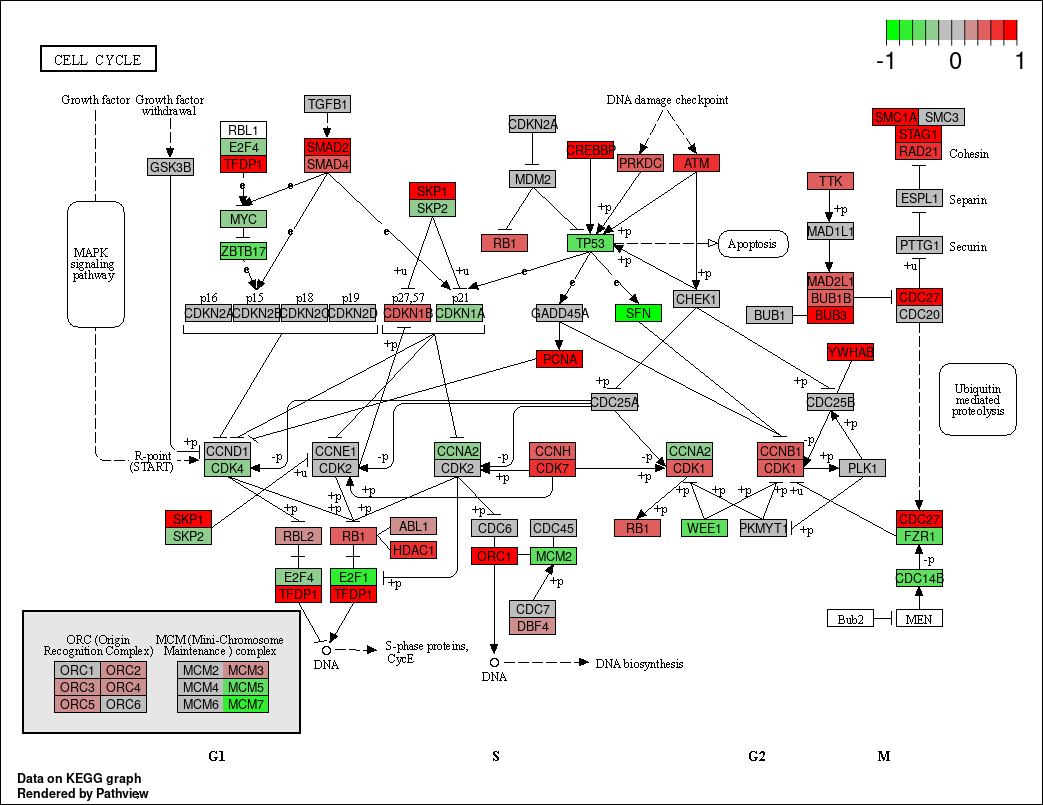
2.2 Graphviz输出形式
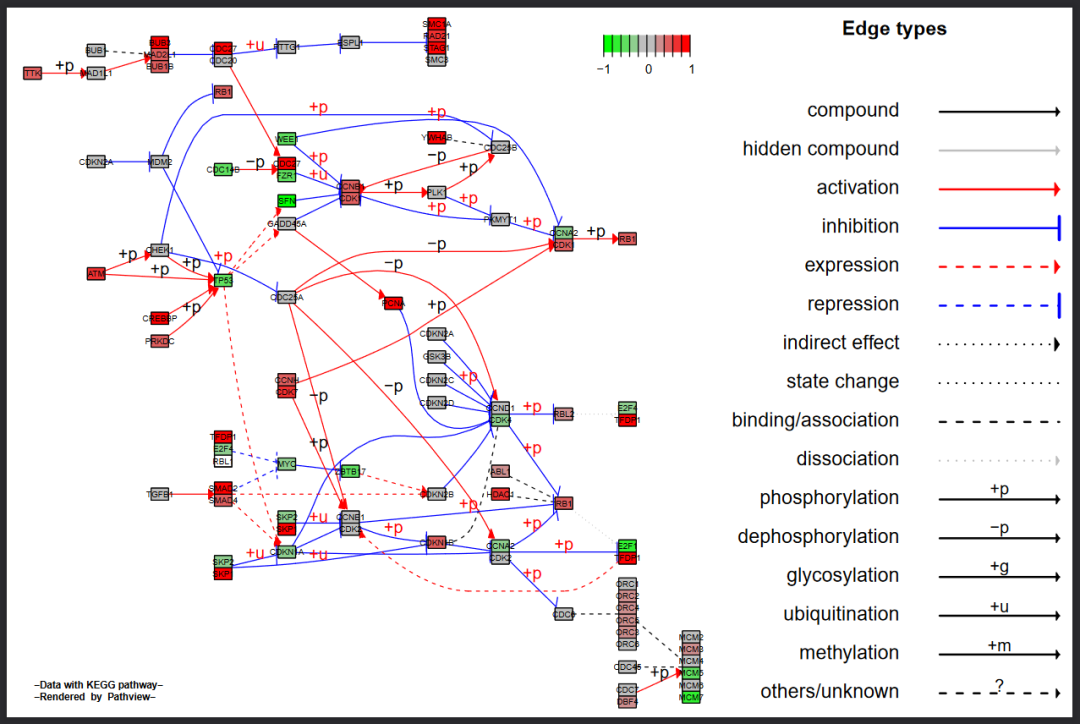
除了最原始的KEGG形式的通路图,我们还可以使用Graphviz视图,设置kegg.native = FALSE即使用Graphvis。后者使用Graphviz引擎布置通路图;因此可以更好地控制节点或边的属性和路径拓扑结构。更重要的是,它的输出是PDF格式的向量图。代码如下:
#Graphviz view: gene data only
pv.out <- pathview(gene.data = gse16873.d[, 1],
pathway.id = demo.paths$sel.paths[i],
species = "hsa",
out.suffix = "gse16873",
kegg.native = FALSE, #设置kegg.native = FALSE即使用Graphvis
sign.pos = demo.paths$spos[i])结果图如下:
在这个例子中,所有的内容同样是被放入到一个图层当中
由于节省空间,默认是只显示了线条的图例
如果相要单独显示节点的图例或是分开图层,同样设置same.layer=FALSE即可
2.2.1 split group
在Graphviz视图中,我们能对图像做更多的控制。比如我们可以将一群节点各自分开;或者将多个基因节点合成一个。这些分离或者聚合的节点会继承之前与未改变的节点间的连接线条。这样就可以得到一个基因或是蛋白质交互网络。只要设置split.group(单个基因节点拆分成数个)或是expand.node(多个基因节点合并)参数即可。
我们先只设置split.group = TRUE来看一下拆分含有多个基因的节点后的效果。
# split group
pv.out1 <- pathview(gene.data = gse16873.d[, 1],
pathway.id = demo.paths$sel.paths[i],
species = "hsa",
out.suffix = "gse16873.split",
kegg.native = F,
sign.pos = demo.paths$spos[i],
split.group = T)
> dim(pv.out1$plot.data.gene)
[1] 92 10结果如下: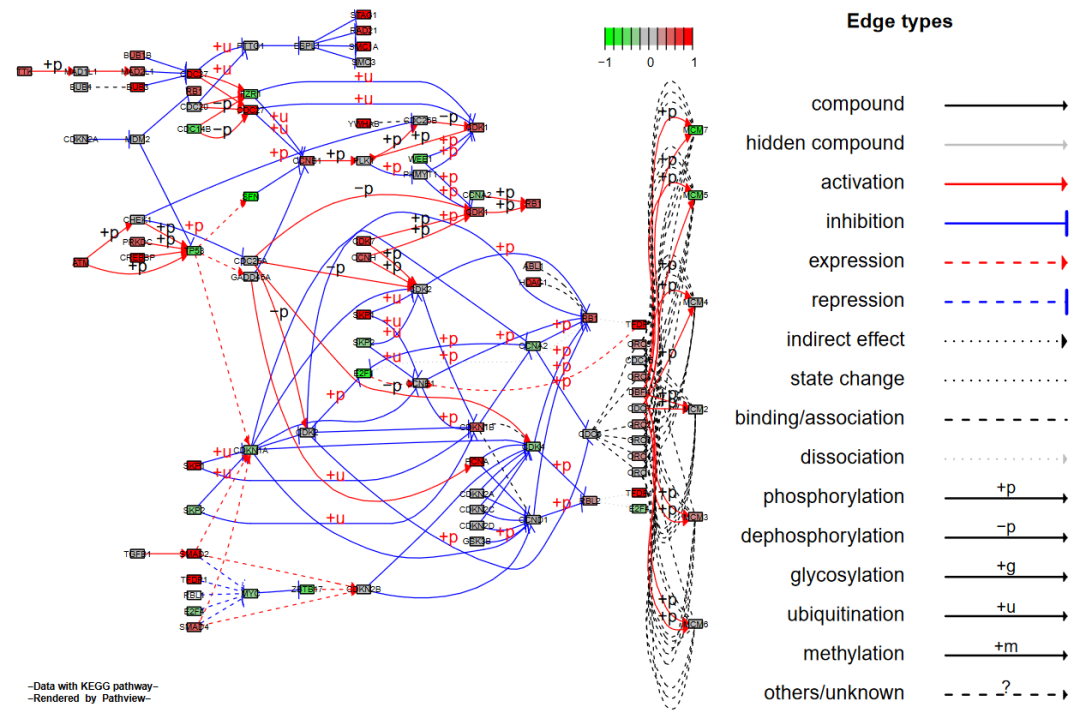
2.2.2 split + expand.node
我们同时设置split.group = TRUE和expand.node = TRUE
# split + expand.node
pv.out2 <- pathview(gene.data = gse16873.d[, 1],
pathway.id = demo.paths$sel.paths[i],
species = "hsa",
out.suffix = "gse16873.split.expanded",
kegg.native = F,
sign.pos = demo.paths$spos[i],
split.group = T,
expand.node = T)
> dim(pv.out2$plot.data.gene)
[1] 126 10结果如下: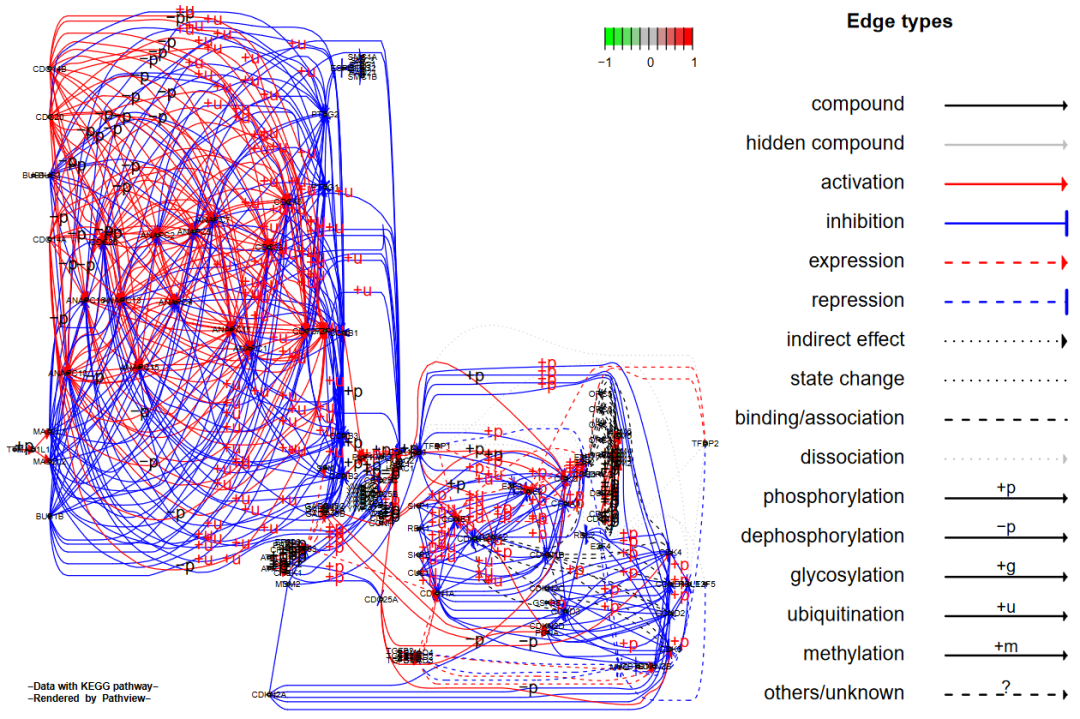
注意在传统的KEGG通路图上,一个基因节点有时包含则多个有类似功能作用的基因或蛋白质,将它们合并到一起是为了图像简洁明了。所以pathview函数默认上不会将节点分开,而是通过总结基因数据再展示到对应节点上。使用者可以通过node.sum参数来定义整合这些在同一节点上的基因的数据的方法。
3. 小结
今天我们主要是对包中给的主函数pathview()做了一个全面的了解。此外我们还学习了Pathview包输出通路图时的两种形式:原始KEGG通路图和Graphviz格式的通路图。最后我们还学习了通路图拆分以及合并节点的方法。














 5796
5796











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









