






问题
kegg Error in clusterProfiler:::kegg_list(“pathway”, species): unused argument (species)
cluserProfile包更新失败
原理
- 超几何分布检验
pathway_pvals <- 1 - phyper(overlap_size - 1, pathway_size,
length(kegg_genes)-pathway_size,
length(genes), lower.tail=TRUE #lower.tail=F 表示结果1-重合程度:
)
# 或者
pathway_pvals <- phyper(overlap_size - 1, pathway_size,
length(kegg_genes)-pathway_size,
length(genes), lower.tail=F)
代码实现
suppressPackageStartupMessages({
library('dplyr')
library('stringr')
library('ggplot2')
})
# 1.0 set
state = 'S01'
celltype = 'Monocytes'
# imput ","; return c contain
split_comma <- function(x){
s <- str_split(x,',')[[1]]
return(s)
}
outp <- file.path('./outcome',celltype,state)
dir.create(outp,recursive = T)
Warning message in dir.create(outp, recursive = T):
“'./outcome/Monocytes/S01' already exists”
# 1.1 imput geneset & referenct
# 1.1.1
kegg_pathways <- readRDS('./KEGG_pathwayFull.RDS')
head(kegg_pathways)
#1.1.2 筛选基因集列表
gene_infop <- file.path('gene_info.txt)
genesets <- read.table(gene_infop)
#genes : 需要的基因列表
genes <- rownames(genesets)[genesets$State == state]
# 1.1.3 kegg gene
kegg_genes <- do.call(c,lapply(kegg_pathways$hgnc_symbol,split_comma)) # 可能有空基因过滤一下
kegg_genes<-kegg_genes[kegg_genes != '']
| pathway_id | pathway_name | entrezgene_id | hgnc_symbol | description |
|---|---|---|---|---|
| <chr> | <chr> | <chr> | <chr> | <chr> |
| hsa01100 | Metabolic pathways - Homo sapiens (human) | NA | Metabolic pathways | |
| hsa01200 | Carbon metabolism - Homo sapiens (human) | NA | Carbon metabolism | |
| hsa01210 | 2-Oxocarboxylic acid metabolism - Homo sapiens (human) | NA | 2-Oxocarboxylic acid metabolism | |
| hsa01212 | Fatty acid metabolism - Homo sapiens (human) | NA | Fatty acid metabolism | |
| hsa01230 | Biosynthesis of amino acids - Homo sapiens (human) | NA | Biosynthesis of amino acids | |
| hsa01232 | Nucleotide metabolism - Homo sapiens (human) | NA | Nucleotide metabolism |
# 1.2.1 calculate p.value & count
kegg_outcome <- data.frame()
for(i in c(1 : nrow(kegg_pathways))){
# 如果通路文件没有基因的情况
if(is.na(kegg_pathways$entrezgene_id[i] ))
{
pathway_size =0
overlap_size = 0
pathway_pvals = 1
ratio = 0
} else{
pathway_genes = split_comma(kegg_pathways$hgnc_symbol[i])
pathway_size <- length(pathway_genes)
overlap_size <- length(intersect(pathway_genes, genes))
ratio = overlap_size / pathway_size
pathway_pvals <- 1 - phyper(overlap_size - 1, pathway_size,
length(kegg_genes)-pathway_size,
length(genes), lower.tail=TRUE #lower.tail=F 表示结果1-重合程度:
)
}
#BH方法,得到FDR值
pvals_adjusted <- p.adjust(pathway_pvals, method="BH")
p <- data.frame(kegg_pathways[i,c(1,2,5)],
count=overlap_size ,
p.adjust = pvals_adjusted,
p.value = pathway_pvals,
ratio =ratio)
if( as.numeric(nrow(kegg_outcome)) == 0){
kegg_outcome <- p
}else{
kegg_outcome <- rbind(kegg_outcome,p)
}
}
write.csv(kegg_outcome,file.path(outp, 'kegg_outcome_all.csv'))
# 1.2.2筛选0.05阈值,并且排序
kegg_outcome_filter <- kegg_outcome %>% filter(p.adjust < 0.05) %>%
arrange(p.adjust,desc(count))
write.csv(kegg_outcome_filter,file.path(outp, 'kegg_outcome_all_filter.csv'))
# 1.3 visualize
# 取前20个
KEGG_top20 <- kegg_outcome_filter[1:20,]
#指定绘图顺序(转换为因子):
KEGG_top20$pathway <- factor(KEGG_top20$description,levels = rev(KEGG_top20$description))
# 换行
swr = function(string, nwrap=25) {
paste(strwrap(string, width=nwrap), collapse="\n")
}
swr = Vectorize(swr)
KEGG_top20$pathway<-swr(KEGG_top20$pathway,25)
#Top20富集数目条形图:
mytheme <-theme(plot.title = element_text(hjust = 0.5,size = 14, face = "bold"),
plot.subtitle= element_text(hjust = 0.5,size = 12),
axis.text.y = element_text(size = 12),
legend.title = element_text(size = 14 ),
legend.text = element_text(size = 12))
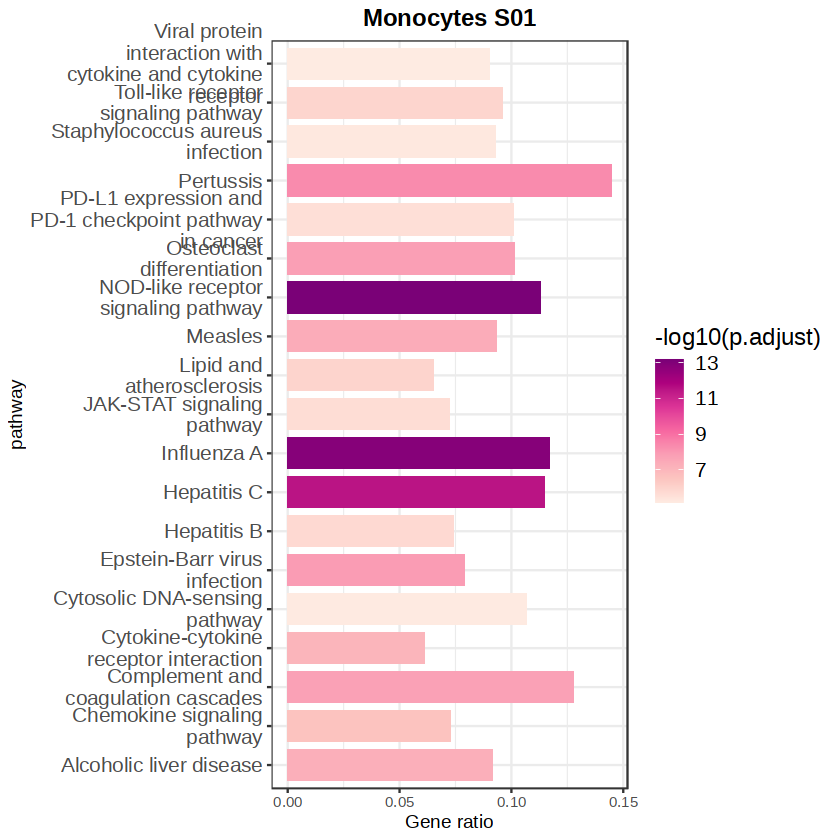
p <- ggplot(data = KEGG_top20[-1,],
aes(x = ratio,
y = pathway,
fill = -log10(p.adjust)))+
# geom_text(aes(label = ratio),vjust = 0.5, hjust = 0)+
scale_fill_distiller(palette = "RdPu",direction = 1) +
geom_bar(stat = "identity",width = 0.8) +
theme_bw() +
labs(x = "Gene ratio",
y = "pathway",
title = paste(celltype, state)) + mytheme
p
ggsave(file.path(outp,"merge.pdf"),width = 6,height = 8 ,limitsize = FALSE) ##ggplot 中直接保存


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









