






 本文详细解释了Seurat中在单细胞数据分析中使用FindIntegrationAnchors函数进行整合的方法,包括anchor的选择、过滤和可靠度评估。同时介绍了FindTransferAnchors函数在细胞定义和联合分析中的应用,以及如何通过计算概率值预测细胞类型和实现空间分布。
本文详细解释了Seurat中在单细胞数据分析中使用FindIntegrationAnchors函数进行整合的方法,包括anchor的选择、过滤和可靠度评估。同时介绍了FindTransferAnchors函数在细胞定义和联合分析中的应用,以及如何通过计算概率值预测细胞类型和实现空间分布。
在单细胞数据中,整合方法是我们用到的最常用的算法,至于函数当然是FindIntegrationAnchors,但是其中有些内容我们需要深入了解一下,
* anchor是怎么找的
* k.anchor、k.filter 、k.score与寻找锚点有什么关系
在这里我们就需要来弄明白整合的一个分析原理。
首先第一步:找anchor
我们在做整合分析的时候,对每个seurat对象都进行了Normalize和FindVariableFeatures,而找到的高变基因就是我们寻找anthor的基础。(默认是2000),当然每个样本找到的高变基因不完全相同,也可以人为指定。对于多个样本的整合,也是重复两个样本的整合,就是对两个的高变基因取交集,进行anthor的查找。(anchor也就是MNN)。
对于anchor寻找的原理,以下图为例:
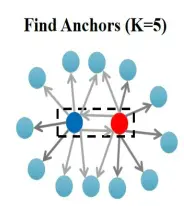
蓝点表示一个样本的细胞,红点表示另外一个样本的细胞,在查找的过程中,蓝点细胞去寻找红点样本中的k个邻居(Seurat里面默认是5),而红点细胞取寻找蓝点样本中的k个邻居,如果此时有双向邻居,则定义为一个anchor(锚点)。这样在数据集中会找到很多的anchor。
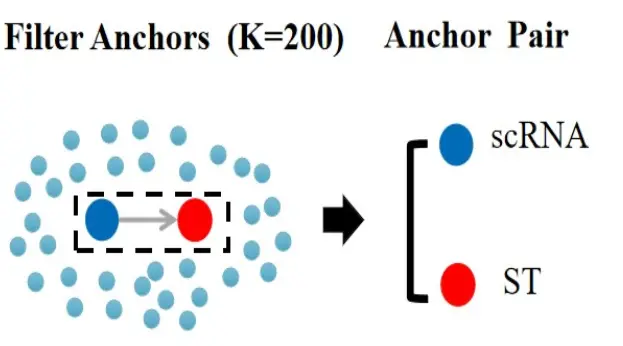
第二步:过滤不可靠的anchor。
不是所有的anthor都可以拿来用的。
在过滤阶段,过滤条件需要放宽,也不要求必须是双向邻居,单向邻居就够了(k.filter: How many neighbors (k) to use when filtering anchors)。这里默认参数是200。200个邻居内所有的anthor都会保留,确定可靠的邻居对。
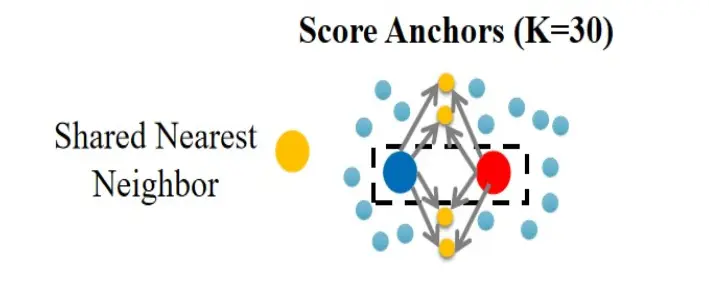
第三步:量化邻居对的可靠程度,采用的是共享邻居的比例。参数是30
说到这里,相信大家都知道了数据整合的最根本原理,由此也知道了control和疾病的样本整合,为什么Seurat会有过拟合的结果。
另外我们来关注一下另外一个函数FindTransferAnchors
这个函数有助于我们做细胞定义、单细胞与空间转录组数据的整合。原理与上面讲到的一致。也是寻找anchors,如下图。
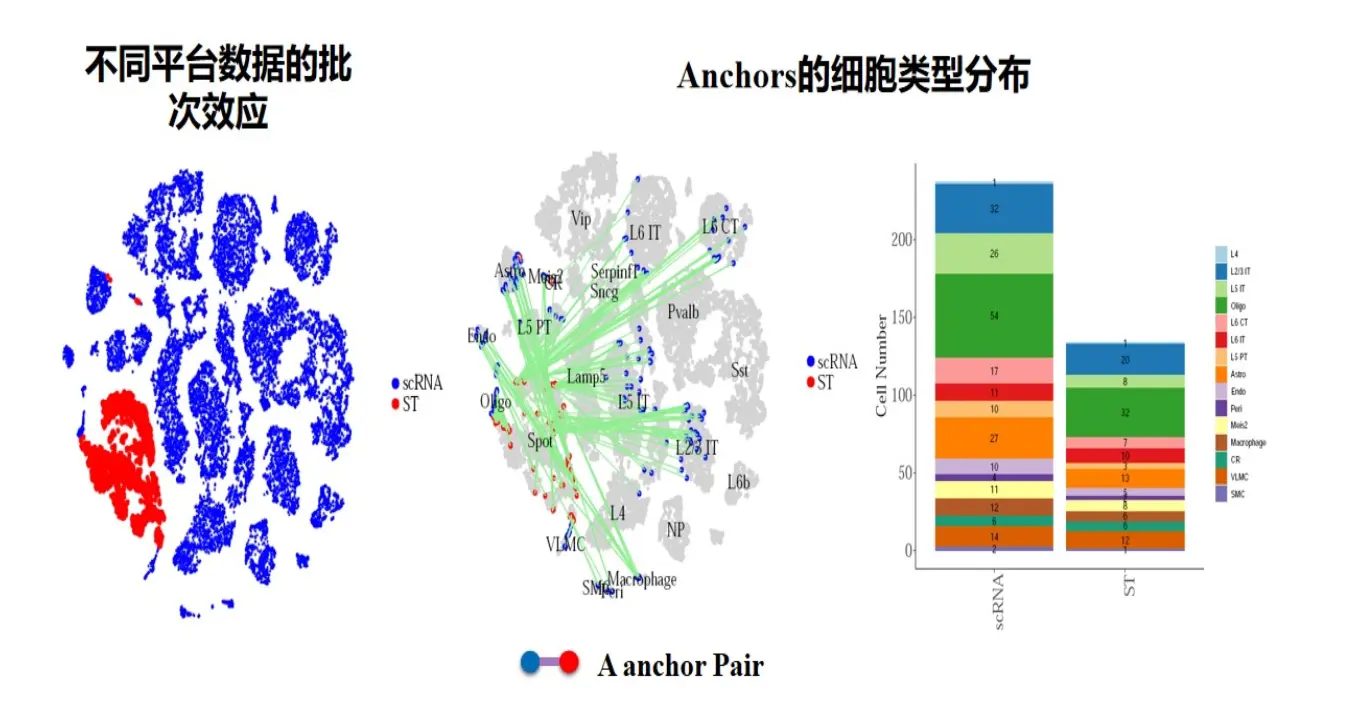
也就是说,我们利用该方法,利用一个定义好的细胞集,通过该函数可以找到相关的anchors,从而识别可能的细胞类型,Seurat在做单细胞和空间的联合分析的时候用到了该方法。但是要注意两点:
(1)单细胞数据和空间数据必须是匹配的
(2)随便找一个单细胞数据都可以得到结果,但没什么用,对应的结果才是正确的。
细胞类型的预测
有了已知的anchors,有一个待预测的cell或者空间spot,我们可以计算出该spot归属于每个anchors的概率,按照细胞类型为单位进行累加,就可以计算出该细胞或者spot属于某种细胞的概率值,概率值加和等于1,具体算法如下:
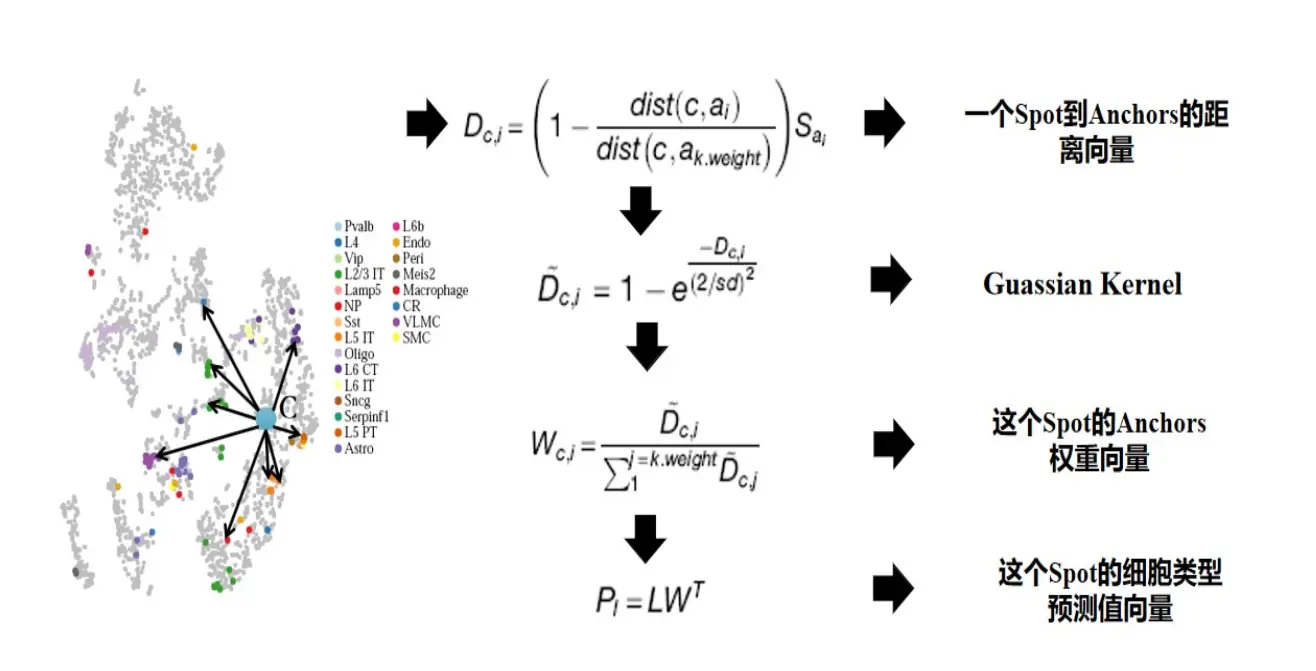
Seurat实现方法:
FindTransferAnchors和TransferData,大家可以多了解一下,真正对单细胞分析成竹在胸。预测结果有如下的表格:
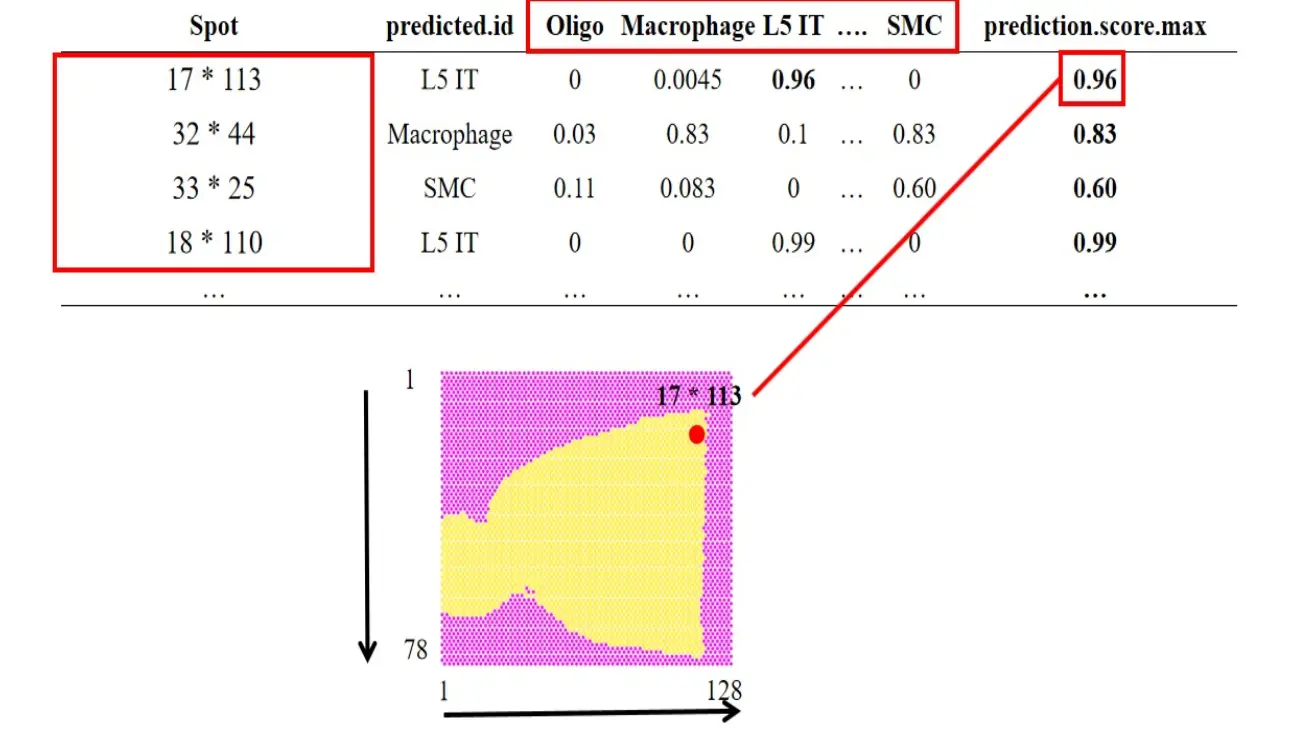
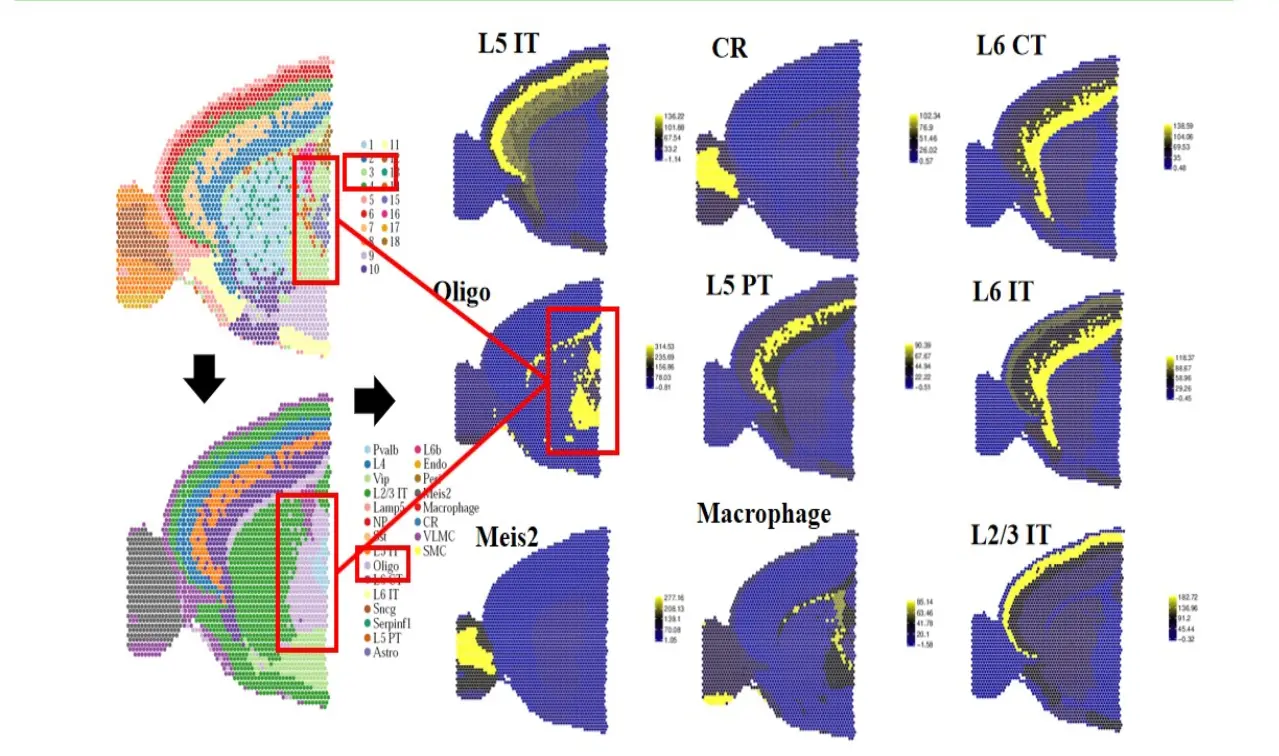
从而实现细胞类型的空间分布:


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









