







–https://satijalab.org/seurat/articles/visiumhd_analysis_vignette
留意更多内容,欢迎关注微信公众号:组学之心
1.数据准备-Seurat
Visium HD 数据是由特定空间排列分布的寡核苷酸序列在 2um x 2um 的网格(bin)中生成的。然而,由于这种分辨率下的数据稀疏,相邻的bin被合并在一起以创建 8um 和 16um 的分辨率。10x 公司建议使用 8um 的合并数据进行分析,但 Seurat 支持同时加载多个合并的数据,并将它们存储在一个对象中作为多个子项目。
接下来是针对小鼠大脑的 Visium HD 数据集分析,下载网址:https://www.10xgenomics.com/datasets/visium-hd-cytassist-gene-expression-libraries-of-mouse-brain-he
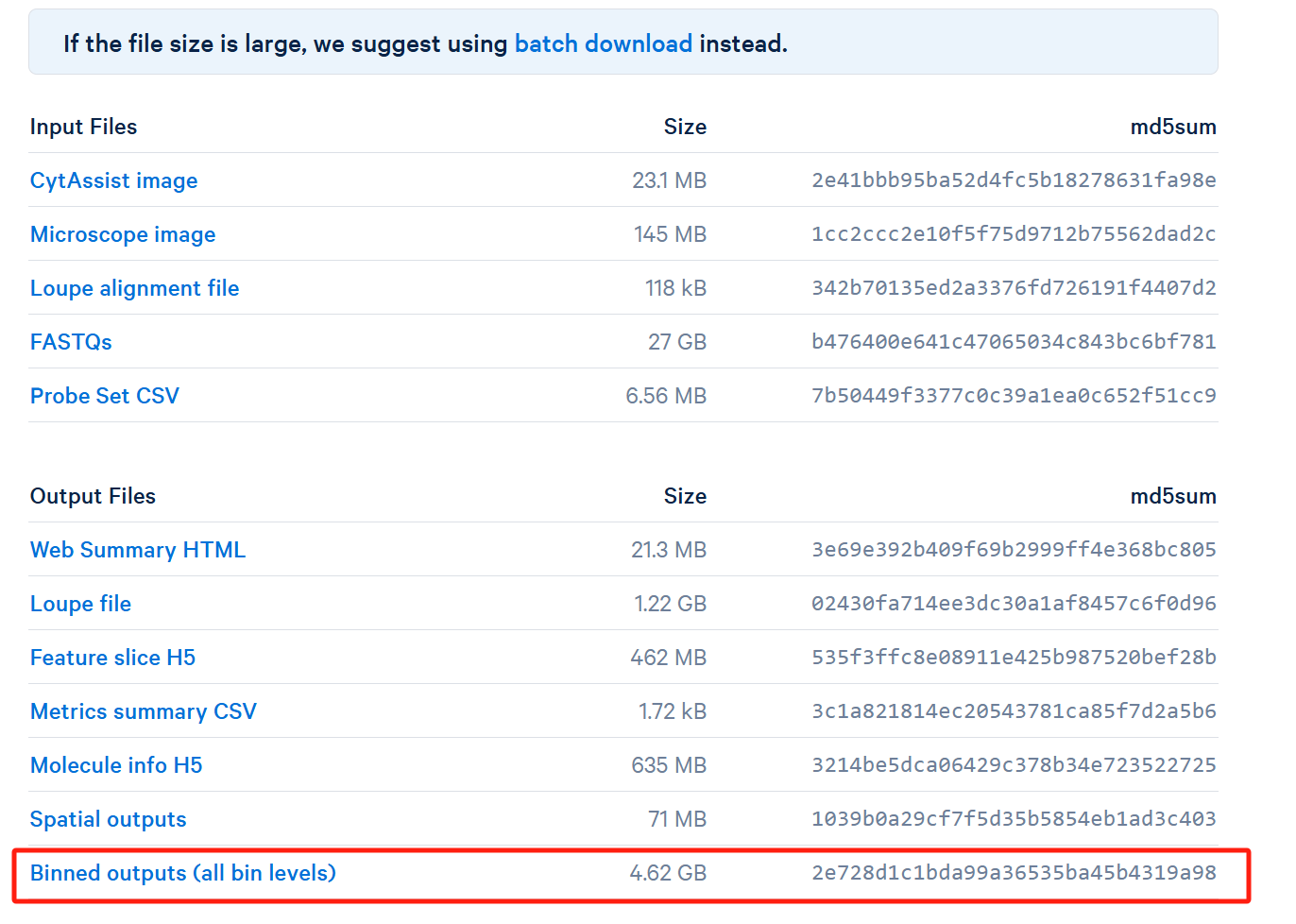
直接下载Binned output即可,下载完后解压得到binned_outputs文件夹,里面的内容长这样:

点开其中一个里面的内容如下:

主要是测序数据:filtered_feature_bc_matrix、raw_feature_bc_matrix和图像数据:spatial
2.聚类降维
2.1 加载Visium HD数据
Load10X_Spatial函数会自动打开mouse_brain文件夹下的binned_outputs,默认情况下会自动读取square_008um和square_016um的测序数据,可以通过bin.size参数来自定义需要读取的数据。Seurat 可以在不同的分析中存储多个分辨率数据。
localdir <- "00practice/VisiumHD/mouse_brain"
hd_data <- Load10X_Spatial(data.dir = localdir, bin.size = c(8, 16))
# Setting default assay changes between 8um and 16um binning
Assays(hd_data)
DefaultAssay(hd_data) <- "Spatial.008um"
Spatial.008um和Spatial.016um存储在hd_data中的assays,可见在8um分辨率下有393543个细胞,19059个基因。
查看细胞中检测到的nCount数目:
vln.plot <- VlnPlot(hd_data, features = "nCount_Spatial.008um", pt.size = 0) + theme(axis.text = element_text(size = 4)) + NoLegend()
count.plot <- SpatialFeaturePlot(hd_data, features = "nCount_Spatial.008um") + theme(legend.position = "right")
vln.plot | count.plot
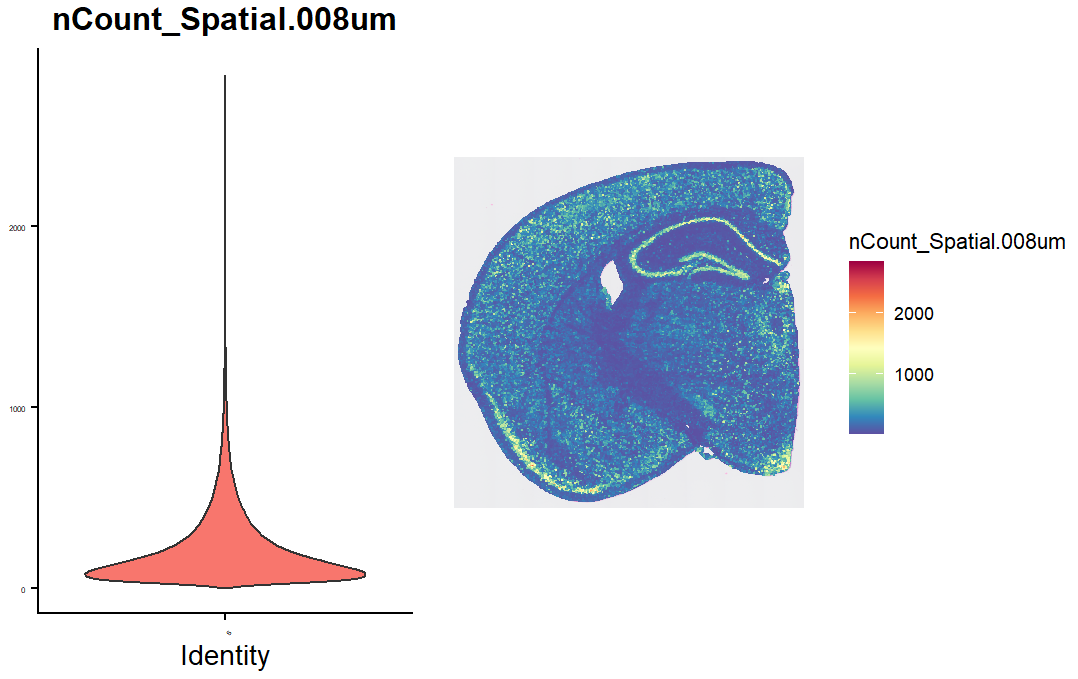
可以看到许多spot的count很少,部分原因是某些组织区域的细胞密度较低
2.2 标准化数据并可视化基因表达情况
# 标准化 8um和16um bins的数据
DefaultAssay(hd_data) <- "Spatial.008um"
hd_data <- NormalizeData(hd_data)
DefaultAssay(hd_data) <- "Spatial.016um"
hd_data <- NormalizeData(hd_data)
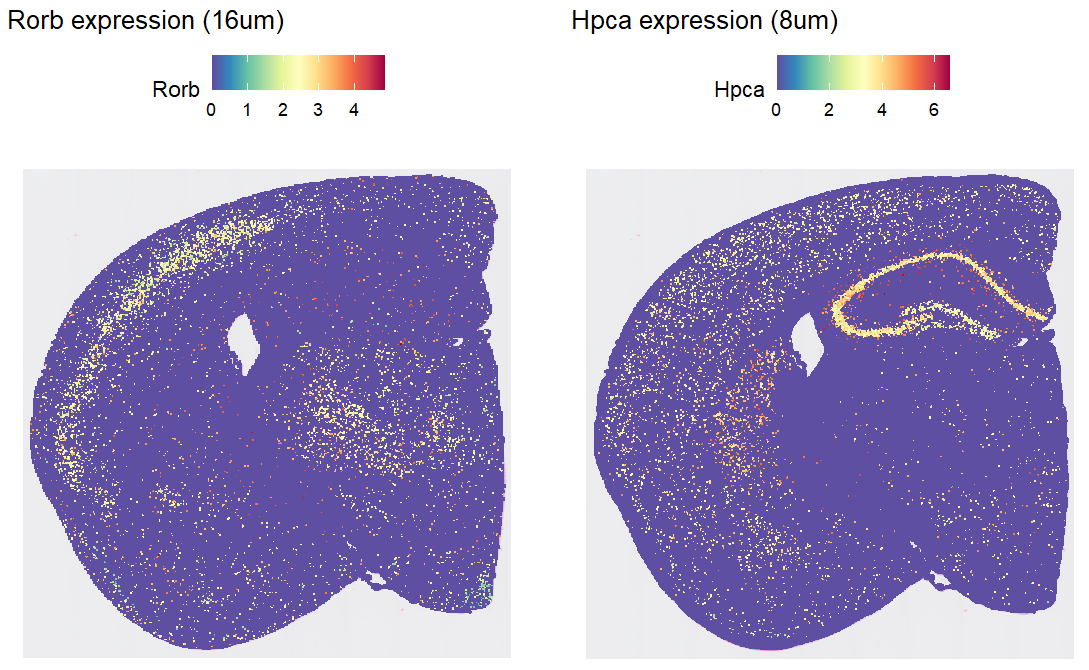
# 在16um分辨率下展现基因表达值的空间分布
DefaultAssay(object) <- "Spatial.016um"
p1 <- SpatialFeaturePlot(object, features = "Rorb") + ggtitle("Rorb expression (16um)")
# 在8um分辨率下展现基因表达值的空间分布
DefaultAssay(object) <- "Spatial.008um"
p2 <- SpatialFeaturePlot(object, features = "Hpca") + ggtitle("Hpca expression (8um)")
p1 | p2
2.3 用sketch-based的方法聚类降维
sketch-based的分析旨在以保护稀有种群的方式对大型数据集进行“subsample”。在这里,seurat首先绘制Visium HD数据集的sketch,对subsample的细胞执行聚类,然后将聚类标签投影回完整的数据集。该方法的论文是https://doi.org/10.1038/s41587-023-01767-y
代码实现如下:
# 用标准化之后的数据进行subsample
DefaultAssay(hd_data) <- "Spatial.008um"
hd_data <- FindVariableFeatures(hd_data)
hd_data <- ScaleData(hd_data)
# 挑选出5000个细胞作为'sketch' assay
hd_data <- SketchData(
object = hd_data,
ncells = 50000,
method = "LeverageScore",
sketched.assay = "sketch"
)
此时的hd_data有三个assay:

接下来对sketch的assay进行聚类降维的操作:
# 切换到sketch的细胞
DefaultAssay(hd_data) <- "sketch"
# 执行聚类的流程代码
hd_data <- FindVariableFeatures(hd_data)
hd_data <- ScaleData(hd_data)
hd_data <- RunPCA(hd_data, assay = "sketch", reduction.name = "pca.sketch")
hd_data <- FindNeighbors(hd_data, assay = "sketch", reduction = "pca.sketch", dims = 1:50)
hd_data <- FindClusters(hd_data, cluster.name = "seurat_cluster.sketched", resolution = 3)
hd_data <- RunUMAP(hd_data, reduction = "pca.sketch", reduction.name = "umap.sketch",
return.model = T, dims = 1:50)
现在可以使用ProjectData函数将聚类标签和从50000个sketch细胞中学习到的降维信息(PCA和UMAP)投影到整个数据集:
hd_data <- ProjectData(
object = hd_data,
assay = "Spatial.008um", #指定投射的全部数据
full.reduction = "full.pca.sketch",
sketched.assay = "sketch",
sketched.reduction = "pca.sketch",
umap.model = "umap.sketch",
dims = 1:50,
refdata = list(seurat_cluster.projected = "seurat_cluster.sketched") #参考数据
)
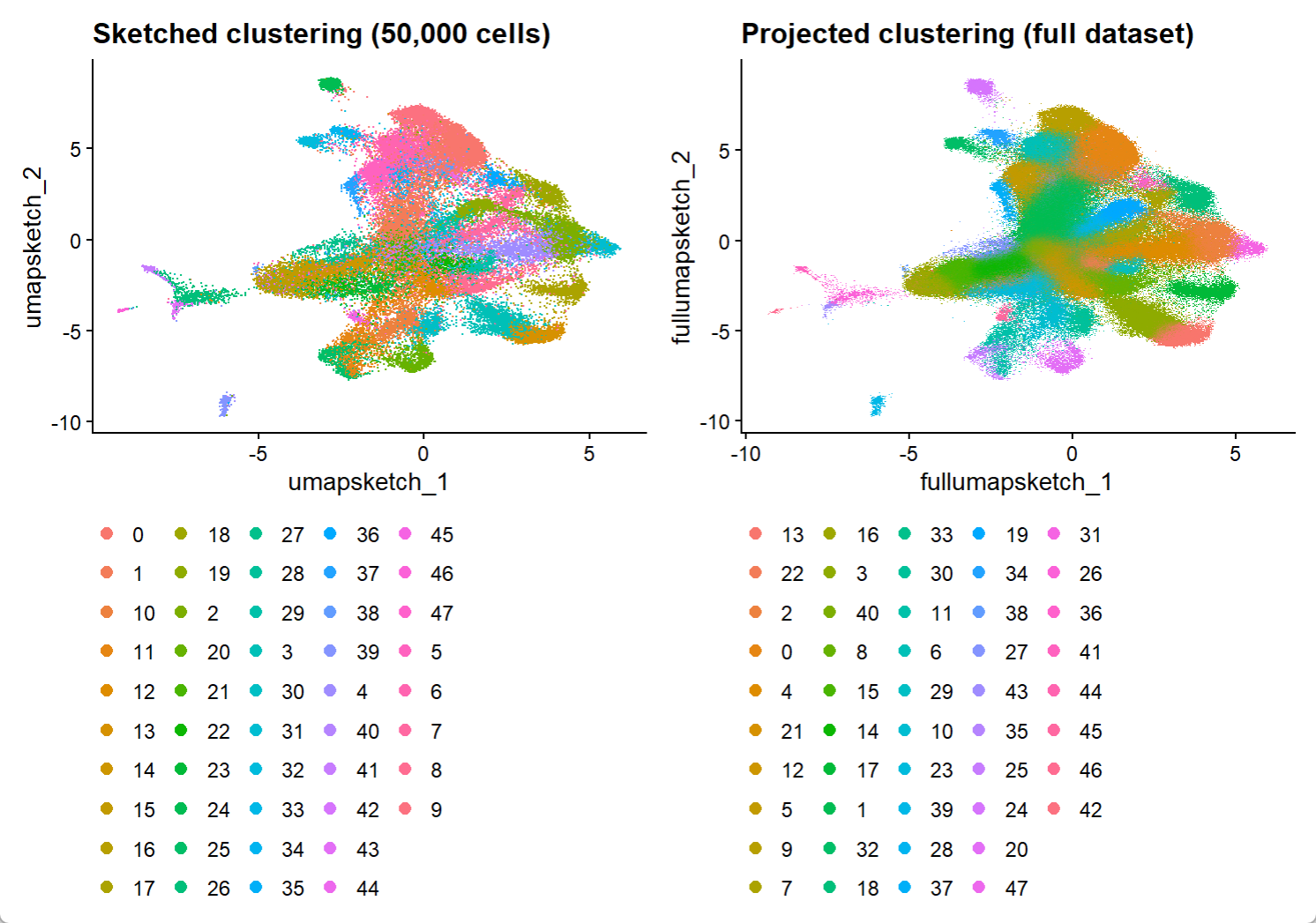
2.4 聚类降维结果的可视化
可视化sketch细胞的聚类结果和整个数据集的投影聚类结果:
DefaultAssay(hd_data) <- "sketch"
Idents(hd_data) <- "seurat_cluster.sketched"
p1 <- DimPlot(hd_data, reduction = "umap.sketch", label = F) + ggtitle("Sketched clustering (50,000 cells)") + theme(legend.position = "bottom")
DefaultAssay(hd_data) <- "Spatial.008um"
Idents(hd_data) <- "seurat_cluster.projected"
p2 <- DimPlot(hd_data, reduction = "full.umap.sketch", label = F) + ggtitle("Projected clustering (full dataset)") + theme(legend.position = "bottom")
p1 | p2
全部clusters在空间上的分布:
SpatialDimPlot(hd_data, label = T, repel = T, label.size = 4)

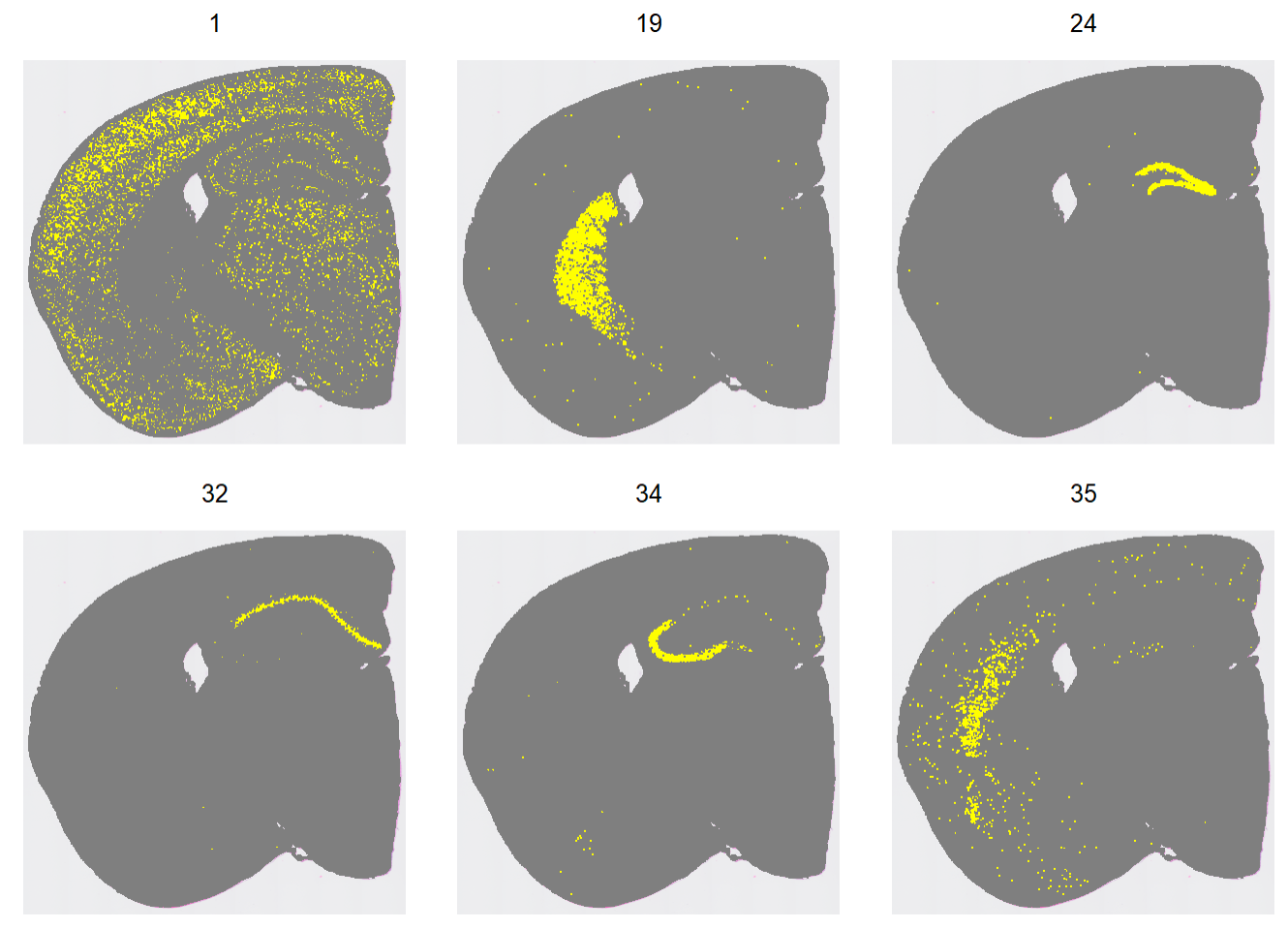
指定clusters在空间上的分布:
Idents(hd_data) <- "seurat_cluster.projected"
cells <- CellsByIdentities(hd_data, idents = c(2, 19, 24, 32, 34, 35))
p <- SpatialDimPlot(hd_data,
cells.highlight = cells[setdiff(names(cells), "NA")],
cols.highlight = c("#FFFF00", "grey50"),
facet.highlight = T, combine = T) + NoLegend()
p
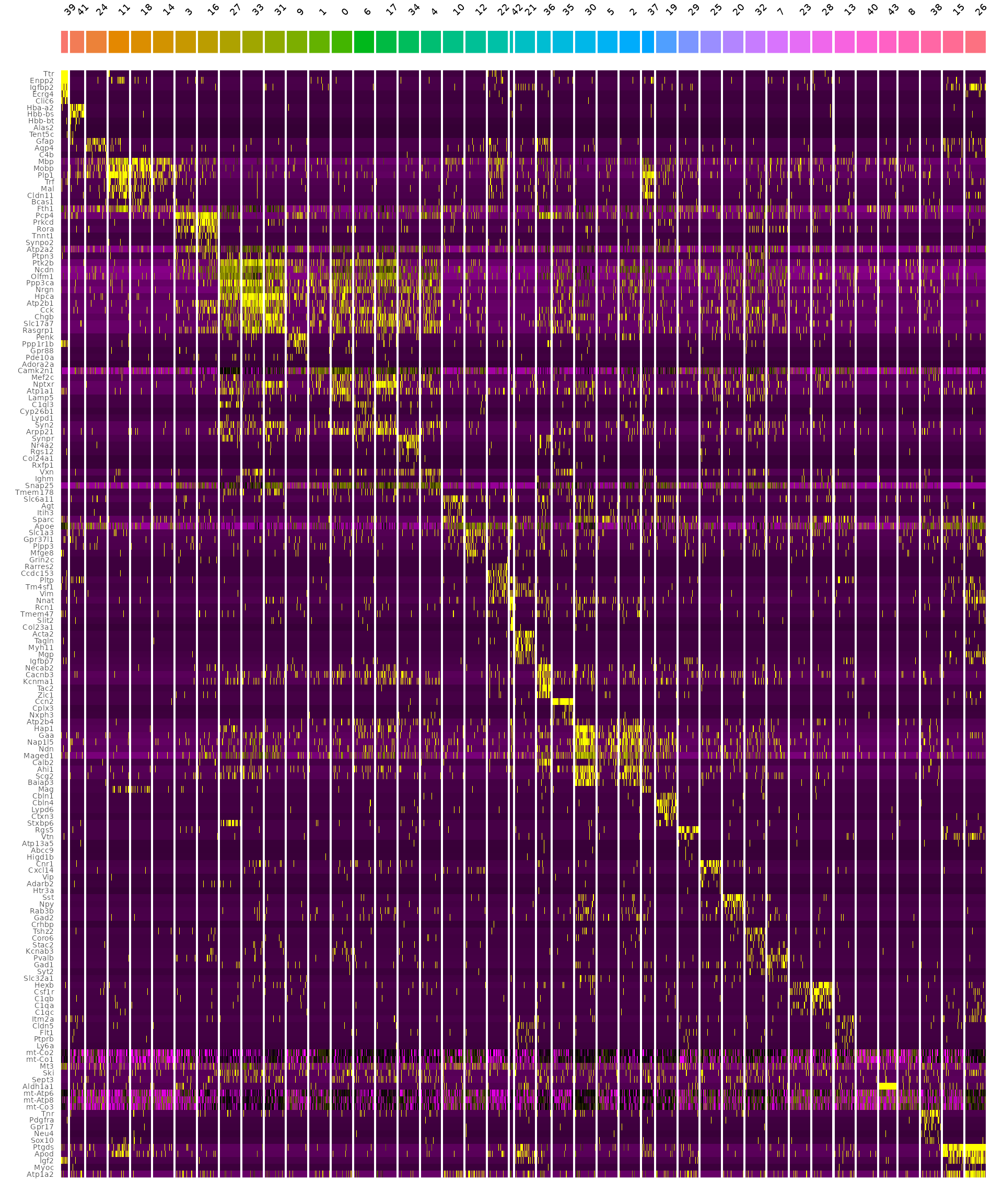
2.5 计算并可视化每个cluster的top基因:
DefaultAssay(hd_data) <- "Spatial.008um"
Idents(hd_data) <- "seurat_cluster.projected"
object_subset <- subset(hd_data, cells = Cells(hd_data[["Spatial.008um"]]), downsample = 1000)
# 根据cluster的相似性进行排序
DefaultAssay(object_subset) <- "Spatial.008um"
Idents(object_subset) <- "seurat_cluster.projected"
object_subset <- BuildClusterTree(object_subset, assay = "Spatial.008um", reduction = "full.pca.sketch", reorder = T)
# 用FindAllMarkers来计算每簇的差异基因
markers <- FindAllMarkers(object_subset, assay = "Spatial.008um", only.pos = TRUE)
markers %>%
group_by(cluster) %>%
dplyr::filter(avg_log2FC > 1) %>%
slice_head(n = 5) %>%
ungroup() -> top5
object_subset <- ScaleData(object_subset, assay = "Spatial.008um", features = top5$gene)
p <- DoHeatmap(object_subset, assay = "Spatial.008um", features = top5$gene, size = 2.5) + theme(axis.text = element_text(size = 5.5)) + NoLegend()
p
这个计算过程需要一段时间…
3.BANKSY-识别空间定义的组织区域
虽然之前的分析独立地考虑了每个细胞,但空间数据不仅可以通过其邻域来定义细胞,还可以通过其更广泛的空间信息来定义细胞。Seurat在这里使用了BANKSY,它对识别和分割组织结构域特别有价值。当进行聚类时,BANKSY通过bins的更宽邻域中基因表达水平的平均值和梯度来增强bins的表达模式。该方法的论文请见:https://doi.org/10.1038/s41588-024-01664-3
3.1 安装BANKSY包
if (!requireNamespace("Banksy", quietly = TRUE)) {
remotes::install_github("prabhakarlab/Banksy@devel")
}
library(SeuratWrappers)
library(Banksy)
在运行BANKSY之前,我们需要考虑两个重要的模型参数:
k_geom:参数定义了每个数据点周围的邻域大小。这个参数决定了一个数据点被认为与哪些其他数据点邻近。k_geom值越大,生成的局部域也越大,模型会更倾向于捕捉数据的全局模式而不是局部细节。较小的k_geom值则相反,会更关注数据的局部变化。
lambda:控制了邻域内数据点的相互影响程度。这个参数决定了邻域中每个点对中心点的影响权重。lambda值越大,生成的域越平滑,邻域内的点之间的相互影响越大,导致更强的空间一致性。较小的lambda值则相反,导致域内各点之间的影响较弱,更能保留原始数据中的局部不一致性。
如果希望模型更关注局部细节,建议使用较小的k_geom和较小的lambda。
如果希望模型更关注全局趋势,建议使用较大的k_geom和较大的lambda。
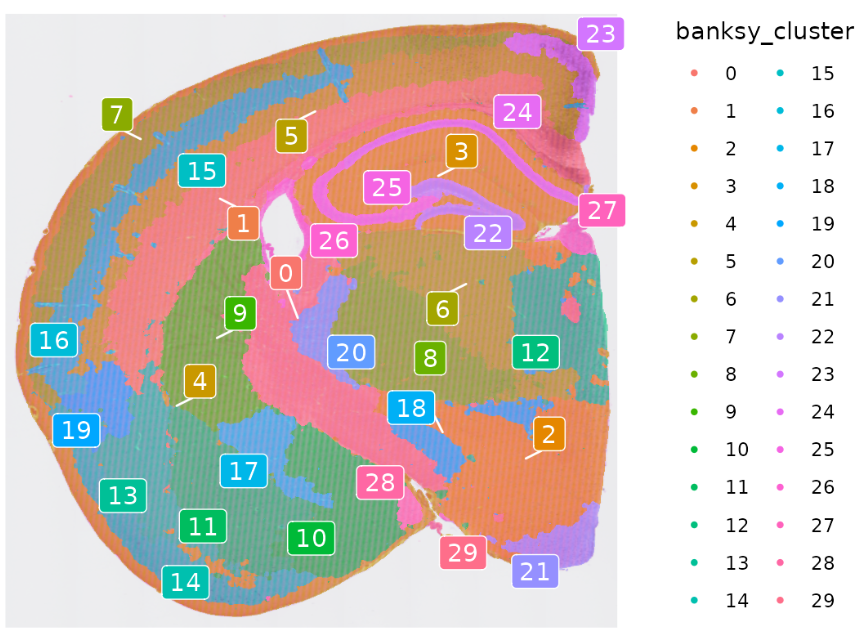
3.2 运行BANKSY执行聚类降维
这一步比较耗运行内存:
hd_data <- RunBanksy(hd_data,
lambda = 0.8, verbose = TRUE,
assay = "Spatial.008um", slot = "data", features = "variable",
k_geom = 50)
DefaultAssay(hd_data) <- "BANKSY"
hd_data <- RunPCA(hd_data, assay = "BANKSY", reduction.name = "pca.banksy", features = rownames(object), npcs = 30)
hd_data <- FindNeighbors(hd_data, reduction = "pca.banksy", dims = 1:30)
hd_data <- FindClusters(hd_data, cluster.name = "banksy_cluster", resolution = 0.5)
Idents(hd_data) <- "banksy_cluster"
p <- SpatialDimPlot(hd_data, group.by = "banksy_cluster", label = T, repel = T, label.size = 4)
p
展示每一簇单独的空间分布情况:
banksy_cells <- CellsByIdentities(hd_data)
p <- SpatialDimPlot(hd_data, cells.highlight = banksy_cells[setdiff(names(banksy_cells), "NA")],
cols.highlight = c("#FFFF00", "grey50"), facet.highlight = T, combine = T) + NoLegend()
p
聚类效果看起来要比前面的方法要好:

4.细分解剖区域
有时候需要分割出感兴趣的区域进行进一步的下游分析。可与使用CreateSegmentation函数从整个数据集中创建一个坐标定义的分割掩模,标记皮层和海马区域,然后使用Overlay函数识别落入该区域的细胞。该数据的坐标列表可在https://www.dropbox.com/scl/fi/qbs3j1alq33f0qz892ub3/cortex-hippocampus_coordinates.csv?rlkey=lsxglb15jhjdrircy9lb6n0rd&dl=0下载,我们可以在使用SpatialDimPlot的交互式=TRUE参数探索自己的数据集时识别这些边界。
cortex.coordinates <- as.data.frame(read.csv("/brahms/lis/visium_hd/final_mouse/cortex-hippocampus_coordinates.csv"))
cortex <- CreateSegmentation(cortex.coordinates)
hd_data[["cortex"]] <- Overlay(hd_data[["slice1.008um"]], cortex)
cortex <- subset(hd_data, cells = Cells(hd_data[["cortex"]]))
下一步将用到cortex Visium HD object
5.与scRNA-seq数据整合分析(去卷积)
这里借助RCTD去卷积方法来整合scRNA-seq数据,它的官方教程网站请见https://raw.githack.com/dmcable/spacexr/master/vignettes/spatial-transcriptomics.html
RCTD以scRNA-seq数据集作为参考,以空间数据集作为查询。使用Allen Brain Atlas的小鼠scRNA-seq数据集作为参考,下载连接:https://www.dropbox.com/scl/fi/r1mixf4eof2cot891n215/allen_scRNAseq_ref.Rds?rlkey=ynr6s6wu1efqsjsu3h40vitt7&dl=0。参考scRNA-seq数据集已减少到200000个细胞(删除了<25个细胞的罕见细胞类型)。
使用cortex Visium HD对象作为空间查询。seurat作者说:“为了提高计算效率,我们绘制了空间查询数据集”,应用RCTD对“sketch”的cortex细胞进行去卷积并对其进行注释,然后将这些注释投影到完整的cortex数据集。
5.1 sketch Visium HD数据集的cortex子集
方法和上面提到的类似
DefaultAssay(cortex) <- "Spatial.008um"
cortex <- FindVariableFeatures(cortex)
cortex <- SketchData(
object = cortex,
ncells = 50000,
method = "LeverageScore",
sketched.assay = "sketch"
)
DefaultAssay(cortex) <- "sketch"
cortex <- ScaleData(cortex)
cortex <- RunPCA(cortex, assay = "sketch", reduction.name = "pca.cortex.sketch", verbose = T)
cortex <- FindNeighbors(cortex, reduction = "pca.cortex.sketch", dims = 1:50)
cortex <- RunUMAP(cortex, reduction = "pca.cortex.sketch", reduction.name = "umap.cortex.sketch", return.model = T, dims = 1:50, verbose = T)
5.2 加载reference scRNA-seq数据集并预处理
ref <- readRDS("00practice/VisiumHD/mouse_brain/allen_scRNAseq_ref.Rds")
Idents(ref) <- "subclass_label"
counts <- ref[["RNA"]]$counts
cluster <- as.factor(ref$subclass_label)
nUMI <- ref$nCount_RNA
levels(cluster) <- gsub("/", "-", levels(cluster))
cluster <- droplevels(cluster)
5.3 开始RCTD分析
# 创建RCTD参考的数据object
reference <- Reference(counts, cluster, nUMI)
counts_hd <- cortex[["sketch"]]$counts
cortex_cells_hd <- colnames(cortex[["sketch"]])
coords <- GetTissueCoordinates(cortex)[cortex_cells_hd, 1:2]
# 创建RCTD参考的查询object
query <- SpatialRNA(coords, counts_hd, colSums(counts_hd))
# 运行RCTD
RCTD <- create.RCTD(query, reference, max_cores = 28)
RCTD <- run.RCTD(RCTD, doublet_mode = "doublet")
# 把输出结果添加到Seurat object中
cortex <- AddMetaData(cortex, metadata = RCTD@results$results_df)
5.4 将sketch的cortex细胞的RCTD标签投影到所有cortex细胞中
cortex$first_type <- as.character(cortex$first_type)
cortex$first_type[is.na(cortex$first_type)] <- "Unknown"
cortex <- ProjectData(
object = cortex,
assay = "Spatial.008um",
full.reduction = "pca.cortex",
sketched.assay = "sketch",
sketched.reduction = "pca.cortex.sketch",
umap.model = "umap.cortex.sketch",
dims = 1:50,
refdata = list(full_first_type = "first_type")
)
我们只对cortex细胞进行了随机对照试验,将所有其他细胞的label设置为“Unknown”:
DefaultAssay(hd_data) <- "Spatial.008um"
hd_data[[]][, "full_first_type"] <- "Unknown"
hd_data$full_first_type[Cells(cortex)] <- cortex$full_first_type[Cells(cortex)]
现在可以在空间上绘制出scRNA-seq数据中细胞类型的空间分布图:
Idents(hd_data) <- "full_first_type"
cells <- CellsByIdentities(hd_data)
excitatory_names <- sort(grep("^L.* CTX", names(cells), value = TRUE))
p <- SpatialDimPlot(hd_data, cells.highlight = cells[excitatory_names], cols.highlight = c("#FFFF00", "grey50"), facet.highlight = T, combine = T, ncol = 4)
p
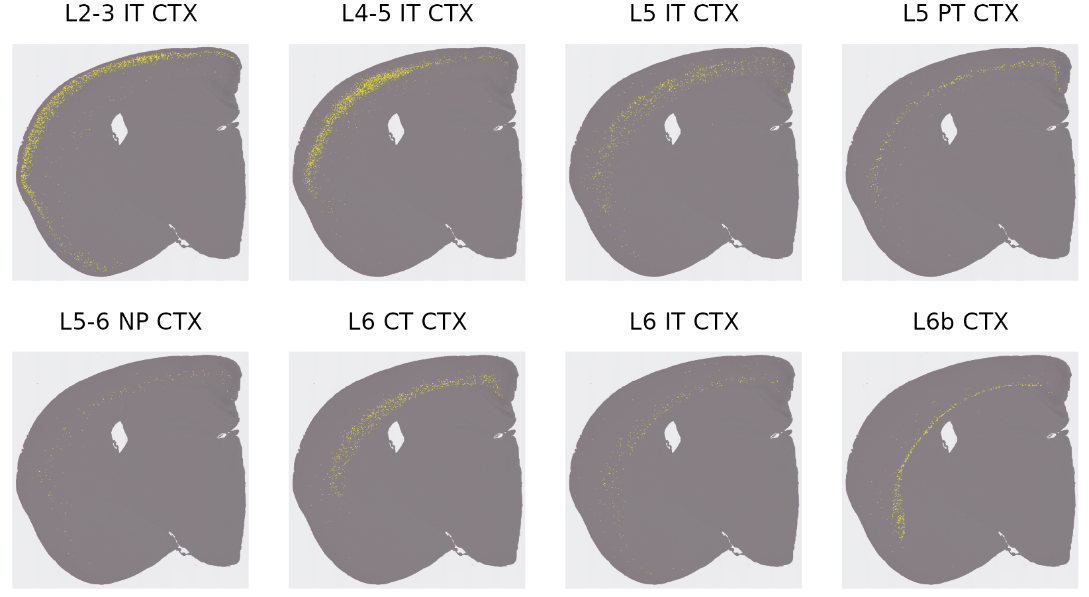
5.5 将RCTD去卷积结果和BANKSY clusters联动
我们现在可以寻找单个bin的scRNA-seq标签与其BANKSY clusters之间的关联。通过查询兴奋性神经元细胞属于哪些空间域,我们可以将对应的BANKSY clusters重命名为神经元层:
plot_cell_types <- function(data, label) {
p <- ggplot(data, aes(x = get(label), y = n, fill = full_first_type)) +
geom_bar(stat = "identity", position = "stack") +
geom_text(aes(label = ifelse(n >= min_count_to_show_label, full_first_type, "")), position = position_stack(vjust = 0.5), size = 2) +
xlab(label) +
ylab("# of Spots") +
ggtitle(paste0("Distribution of Cell Types across ", label)) +
theme_minimal()
}
cell_type_banksy_counts <- hd_data[[]] %>%
dplyr::filter(full_first_type %in% excitatory_names) %>%
dplyr::count(full_first_type, banksy_cluster)
min_count_to_show_label <- 20
p <- plot_cell_types(cell_type_banksy_counts, "banksy_cluster")
p
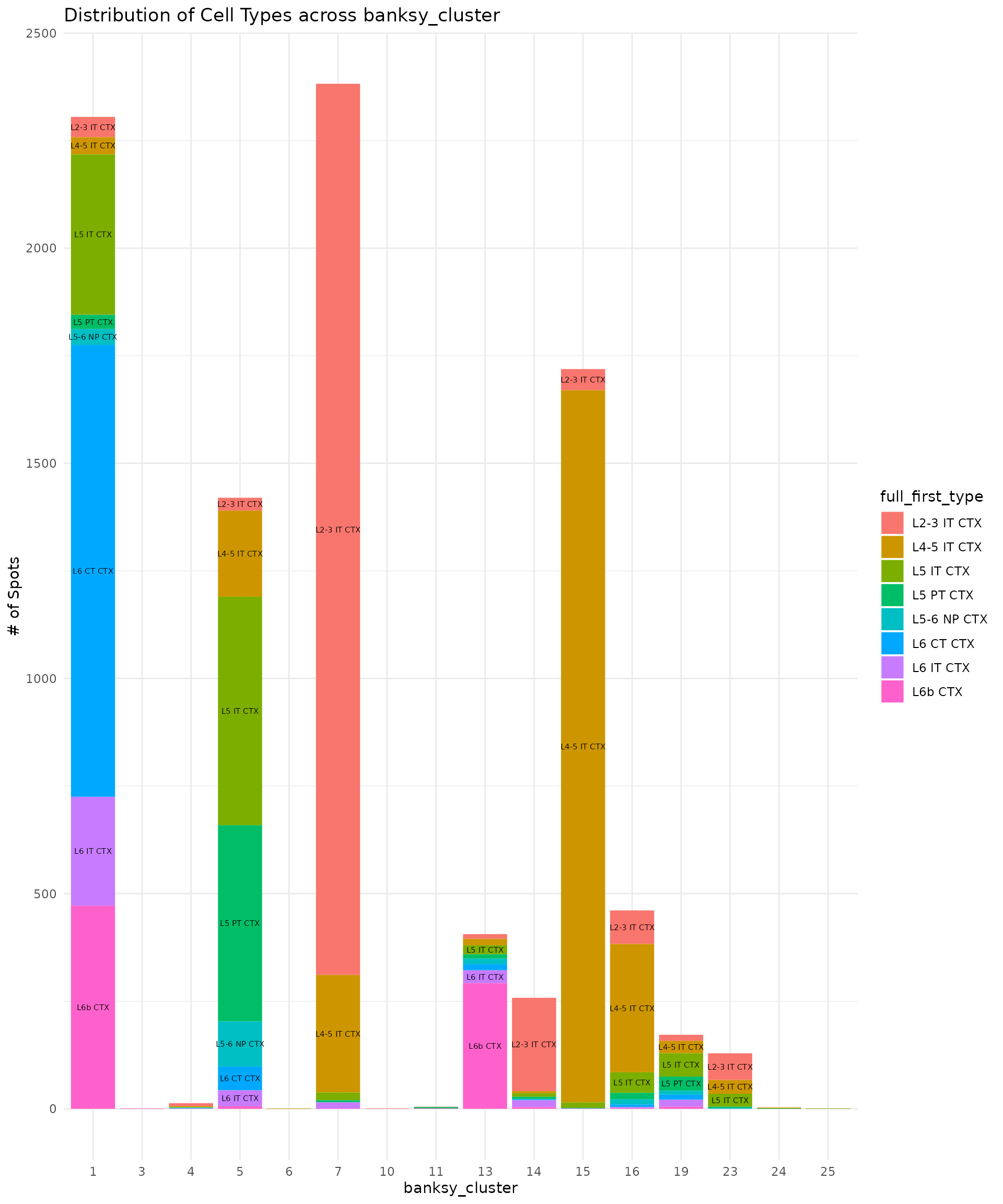
基于这个图,我们现在可以将细胞分配到单个神经元层。
Idents(hd_data) <- "banksy_cluster"
hd_data$layer_id <- "Unknown"
hd_data$layer_id[WhichCells(hd_data, idents = c(7))] <- "Layer 2/3"
hd_data$layer_id[WhichCells(hd_data, idents = c(15))] <- "Layer 4"
hd_data$layer_id[WhichCells(hd_data, idents = c(5))] <- "Layer 5"
hd_data$layer_id[WhichCells(hd_data, idents = c(1))] <- "Layer 6"
最后,我们可以可视化其他细胞类型的空间分布,并查询它们属于哪个皮层。例如,与兴奋性神经元相比,皮层中的抑制性(GABA能)中间神经元在空间上并不局限于单个层。Seurat作者提到之前他们的研究发现SST和PV中间神经元类别往往局限于4-6层,而VIP和Lamp5中间神经元往往位于2/3层。这些结果是基于一种原位成像技术,该技术可以捕捉单细胞的轮廓。那是否可以在基于Visium HD斑点的数据中找到相同的结果呢?
# 把ID设置为RCTD label
Idents(hd_data) <- "full_first_type"
# 可视化4种中间神经元亚型的分布
inhibitory_names <- c("Sst", "Pvalb", "Vip", "Lamp5")
cell_ids <- CellsByIdentities(hd_data, idents = inhibitory_names)
p <- SpatialDimPlot(hd_data, cells.highlight = cell_ids, cols.highlight = c("#FFFF00", "grey50"), facet.highlight = T, combine = T, ncol = 4)
p
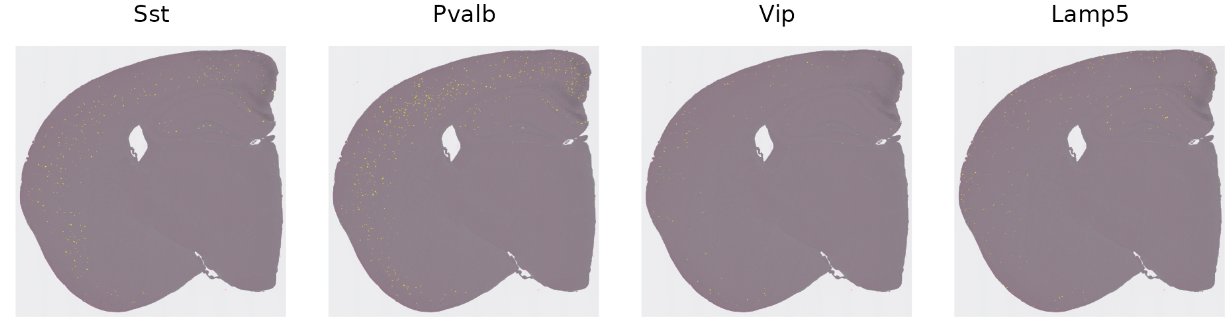
画条形图来展现目标细胞类型的比例:
# 把ID设置为RCTD label
layer_table <- table(hd_data$full_first_type, hd_data$layer_id)[inhibitory_names, 1:4]
neuron_props <- reshape2::melt(prop.table(layer_table), margin = 1)
ggplot(neuron_props, aes(x = Var1, y = value, fill = Var2)) +
geom_bar(stat = "identity", position = "fill") +
labs(x = "Cell type", y = "Proportion", fill = "Layer") +
theme_classic()
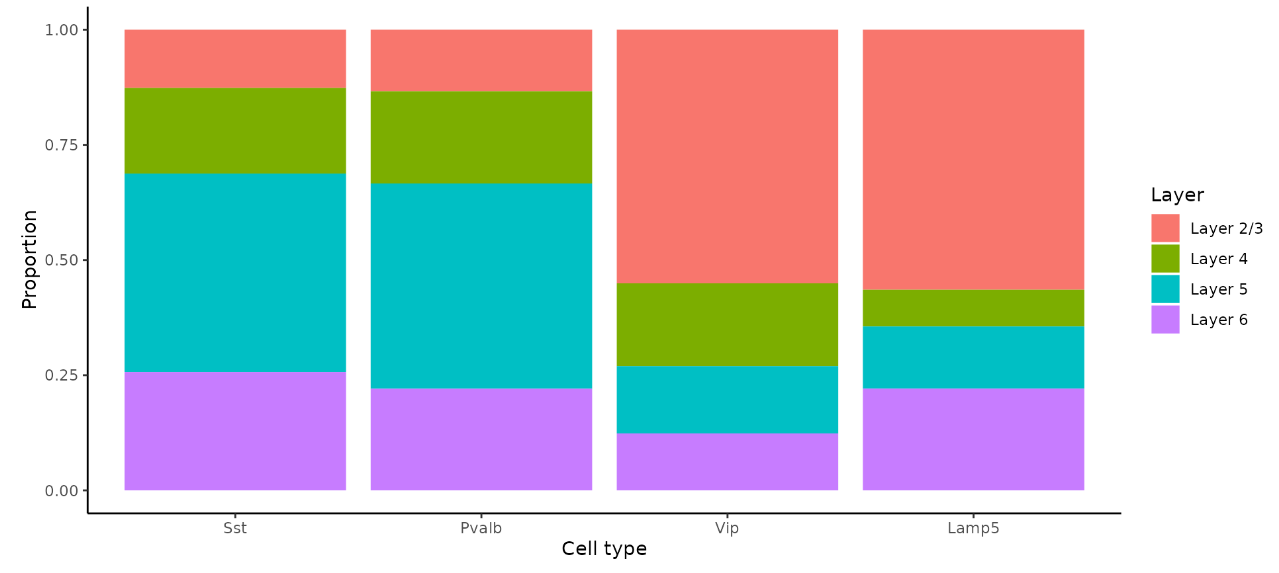
可见,在Visium HD数据集中重述了先前在原位成像数据中发现的相同发现。表明,尽管Visium HD的8um bins不能代表真正的单细胞分辨率,但它能够准确定位scRNA-seq定义的细胞类型。
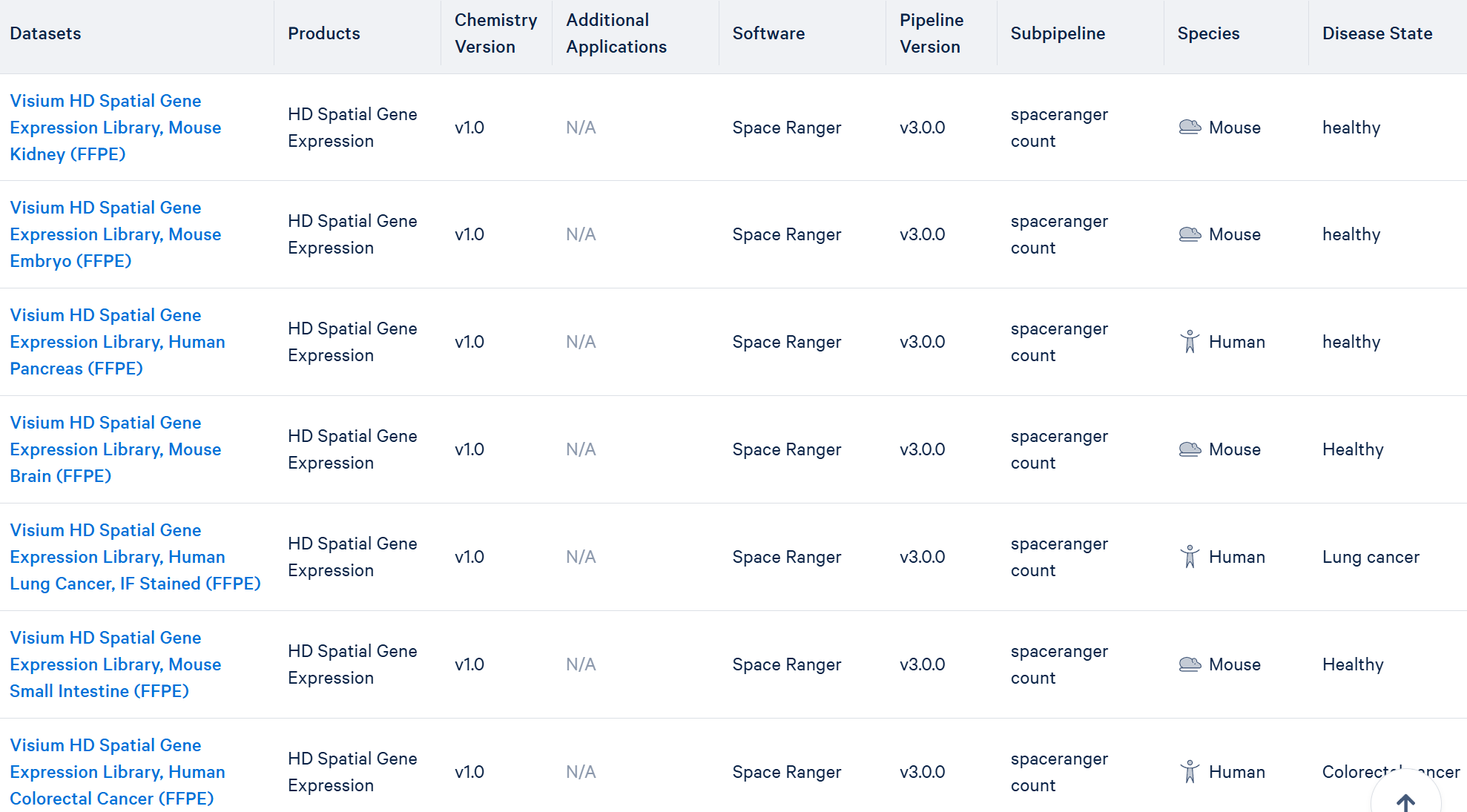
10x的数据集官网目前还提供了其它6种HD的数据:



















 643
643

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











