







自学R语言计算宏基因组显著性+Gephi构建微生物群落的共线网络,为防止后面忘记做个笔记,附赠R代码,以及自学的参考视频/公众号等。
目录
1、R语言求显著性
1.1 计算文件准备
表1:微生物相对丰度数据(.csv文件,对应代码的文件名下面标红的地方)
# ==================== 数据读取与预处理 ====================
cat("正在读取数据...\n")
otu <- read.csv("C:/R/1-1/read-yuxi.csv", row.names = 1, check.names = FALSE)
![]()
1.2 代码
参考这篇公众号:
deepseek根据我的文件位置和文件名为我修改的下面代码,建议按照自己的需求发给AI叫他帮忙改一下,哪里报错就在发给ai叫他改。
# ==================== 初始设置 ====================
# 设置R包安装路径
dir.create("C:/R/packages", showWarnings = FALSE)
.libPaths("C:/R/packages")
# 解决冲突(必须在加载包之前运行!)
conflicts_prefer(dplyr::filter) # 声明优先使用 dplyr 的 filter
# 安装必要包(如果尚未安装)
if (!require("igraph")) install.packages("igraph")
if (!require("psych")) install.packages("psych")
if (!require("tidyverse")) install.packages("tidyverse")
library(igraph)
library(psych)
library(tidyverse)
# 获取当前时间戳
time_stamp <- format(Sys.time(), "%Y%m%d_%H%M%S")
# ==================== 数据读取与预处理 ====================
cat("正在读取数据...\n")
otu <- read.csv("C:/R/1-1/read-yuxi.csv", row.names = 1, check.names = FALSE)
# 过滤低丰度微生物(总丰度≥0%)
otu_rowsums <- rowSums(otu)
otu_filtered <- otu[otu_rowsums >= 0, ]
# 过滤低频出现OTU(在≥1个样本中出现)
otu_binary <- otu_filtered
otu_binary[otu_binary > 0] <- 1
otu_final <- otu_filtered[rowSums(otu_binary) >= 1, ]
# ==================== 相关性分析 ====================
cat("正在进行相关性分析...\n")
cor_result <- corr.test(
t(otu_final),
method = "spearman",
adjust = "fdr",
alpha = 0.05
)
# 提取并过滤相关性矩阵
cor_matrix <- cor_result$r
p_matrix <- cor_result$p
cor_matrix[p_matrix > 0.05 | abs(cor_matrix) < 0.7] <- 0
diag(cor_matrix) <- 0
# 保存相关性矩阵
cor_file <- paste0("C:/R/1-1/相关性矩阵_玉溪_", time_stamp, ".csv")
write.csv(cor_matrix, file = cor_file)
cat("已保存相关性矩阵到:", cor_file, "\n")
# ==================== 网络构建 ====================
cat("正在构建微生物网络...\n")
weighted_matrix <- cor_matrix * p_matrix
diag(weighted_matrix) <- 0
# 构建网络
g <- graph.adjacency(
weighted_matrix,
mode = "undirected",
weighted = TRUE,
diag = FALSE
)
# 简化网络
g <- simplify(g)
# 删除孤立节点
g <- delete_vertices(g, which(degree(g) == 0))
# 设置边属性
E(g)$correlation <- E(g)$weight
E(g)$weight <- abs(E(g)$weight)
E(g)$direction <- ifelse(E(g)$correlation > 0, 1, -1)
# ==================== 网络分析 ====================
cat("正在进行网络分析...\n")
# 生成节点属性表(直接包含数字ID)
node_attributes <- data.frame(
ID = seq_len(vcount(g)), # 关键点:直接生成数字ID
OTU = V(g)$name, # 保留原始OTU名称
Degree = degree(g),
Betweenness = betweenness(g, weights = NA),
Closeness = closeness(g, weights = NA)
)
# 生成边属性表(直接使用数字ID)
edge_attributes <- data.frame(
Source = match(ends(g, es = E(g))[, 1], V(g)$name), # 起点ID
Target = match(ends(g, es = E(g))[, 2], V(g)$name), # 终点ID
Weight = abs(E(g)$correlation), # 权重绝对值
Correlation = E(g)$correlation, # 原始相关系数
Direction = ifelse(E(g)$correlation > 0, "Positive", "Negative")
) %>%
filter(!is.na(Source) & !is.na(Target)) %>% # 删除无效边
filter(Source != Target) # 删除自环
# 保存节点属性
node_file <- paste0("C:/R/1-1/节点属性_玉溪_", time_stamp, ".csv")
write.csv(node_attributes, file = node_file, row.names = FALSE, fileEncoding = "UTF-8")
cat("已保存节点属性到:", node_file, "\n")
# 保存边属性
edge_file <- paste0("C:/R/1-1/边属性_玉溪_", time_stamp, ".csv")
write.csv(edge_attributes, file = edge_file, row.names = FALSE, fileEncoding = "UTF-8")
cat("已保存边属性到:", edge_file, "\n")
# 保存网络图(GraphML格式,兼容Gephi)
graph_file <- paste0("C:/R/1-1/微生物网络_玉溪_", time_stamp, ".graphml")
write_graph(g, file = graph_file, format = "graphml")
cat("已保存网络图到:", graph_file, "\n")
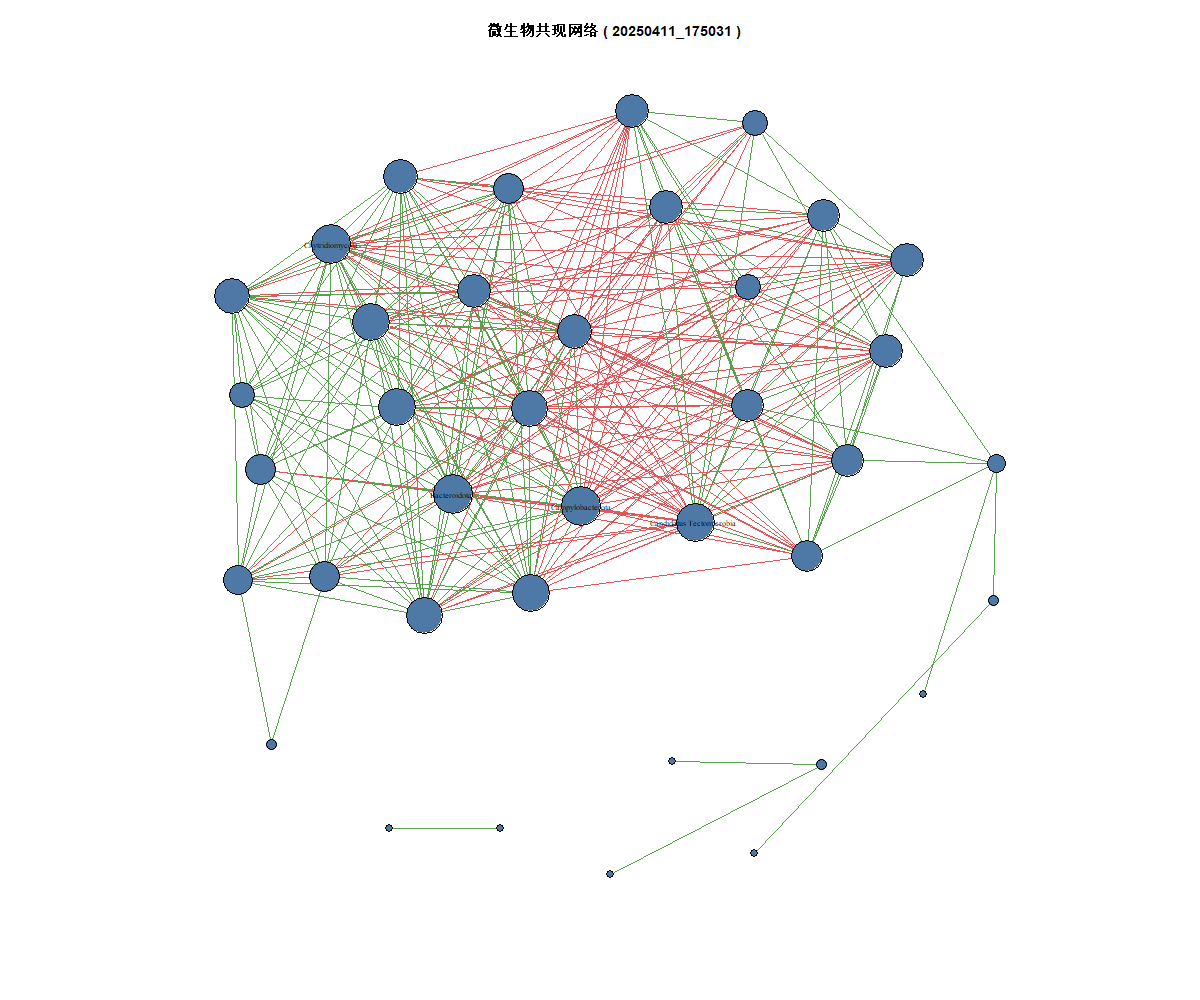
# ==================== 可视化 ====================
cat("正在生成可视化图形...\n")
V(g)$size <- sqrt(degree(g)) * 2
V(g)$color <- "#4E79A7"
E(g)$color <- ifelse(E(g)$direction > 0, "#59A14F", "#E15759")
plot_file <- paste0("C:/R/1-1/网络可视化_玉溪_", time_stamp, ".png")
png(plot_file, width = 1200, height = 1000)
set.seed(123)
plot(g,
layout = layout_with_fr(g),
vertex.label = ifelse(degree(g) > quantile(degree(g), 0.9), V(g)$name, NA),
vertex.label.cex = 0.7,
vertex.label.color = "black",
edge.width = E(g)$weight * 2,
main = paste("微生物共现网络 (", time_stamp, ")")
)
dev.off()
cat("已保存网络可视化图到:", plot_file, "\n")
# ==================== 分析报告 ====================
cat("\n========== 分析结果摘要 ==========\n")
cat("节点数量(OTUs):", vcount(g), "\n")
cat("边数量(相互作用):", ecount(g), "\n")
cat("正相关边比例:", round(mean(E(g)$direction > 0)*100, 1), "%\n")
cat("平均节点度:", round(mean(degree(g)), 1), "\n")
cat("网络直径:", diameter(g, weights = NA), "\n")
cat("平均路径长度:", round(mean_distance(g, weights = NA), 2), "\n")
cat("模块度:", round(modularity(cluster_louvain(g)), 3), "\n")
# 保存分析摘要
summary_file <- paste0("C:/R/1-1/分析摘要_玉溪_", time_stamp, ".txt")
sink(summary_file)
cat("微生物网络分析结果摘要\n")
cat("分析时间:", format(Sys.time(), "%Y年%m月%d日 %H:%M:%S"), "\n\n")
cat("基本参数:\n")
cat("节点数量(OTUs):", vcount(g), "\n")
cat("边数量(相互作用):", ecount(g), "\n")
cat("正相关边比例:", round(mean(E(g)$direction > 0)*100, 1), "%\n\n")
cat("拓扑特征:\n")
cat("平均节点度:", round(mean(degree(g)), 1), "\n")
cat("网络直径:", diameter(g, weights = NA), "\n")
cat("平均路径长度:", round(mean_distance(g, weights = NA), 2), "\n")
cat("模块度:", round(modularity(cluster_louvain(g)), 3), "\n")
sink()
cat("已保存分析摘要到:", summary_file, "\n")
cat("\n========== 分析完成 ==========\n")
如果想要在文章里面形成图片预览可以在最后加上这段代码
# ==================== 交互式预览 ====================
cat("正在生成交互式预览...\n")
# 设置预览参数
V(g)$label <- ifelse(degree(g) > quantile(degree(g), 0.8), V(g)$name, NA)
V(g)$label.cex <- 0.6
V(g)$label.color <- "black"
V(g)$frame.color <- NA
# 创建预览图
set.seed(123)
plot(g,
layout = layout_with_fr(g),
vertex.size = sqrt(degree(g)) * 2,
vertex.color = adjustcolor("#4E79A7", alpha.f = 0.8),
edge.color = ifelse(E(g)$direction > 0,
adjustcolor("#59A14F", alpha.f = 0.5),
adjustcolor("#E15759", alpha.f = 0.5)),
edge.width = abs(E(g)$correlation) * 3,
edge.arrow.size = 0,
main = paste("微生物共现网络预览 (", time_stamp, ")"),
sub = paste("节点:", vcount(g), "| 边:", ecount(g),
"| 正相关:", round(mean(E(g)$direction > 0)*100, 1), "%")
)
# 添加图例
legend("topleft",
legend = c("正相关", "负相关"),
col = c("#59A14F", "#E15759"),
lty = 1,
lwd = 3,
bty = "n",
cex = 0.8)
# 添加度分布直方图作为内嵌图
par(fig = c(0.7, 0.95, 0.7, 0.95), new = TRUE, mar = c(2, 2, 1, 1))
hist(degree(g),
breaks = 20,
col = "#4E79A7",
main = "",
xlab = "节点度",
ylab = "频数")
box()
1.3 分析成功
保存出来的文件就是这样子的了(文件名在代码中找到中文的地方改就行了,建议只改(大理)两个字)。为了防止多次修改分不清文件,所以文件保存的时候自带了保存日期和时间。

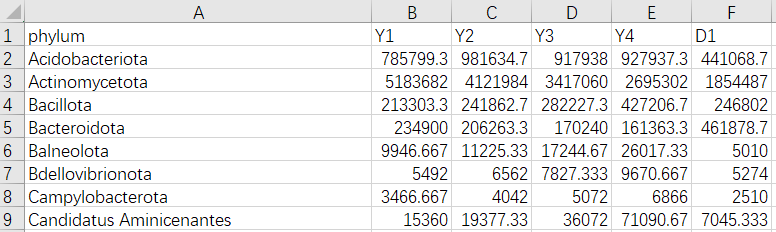
1、相关性矩阵文件
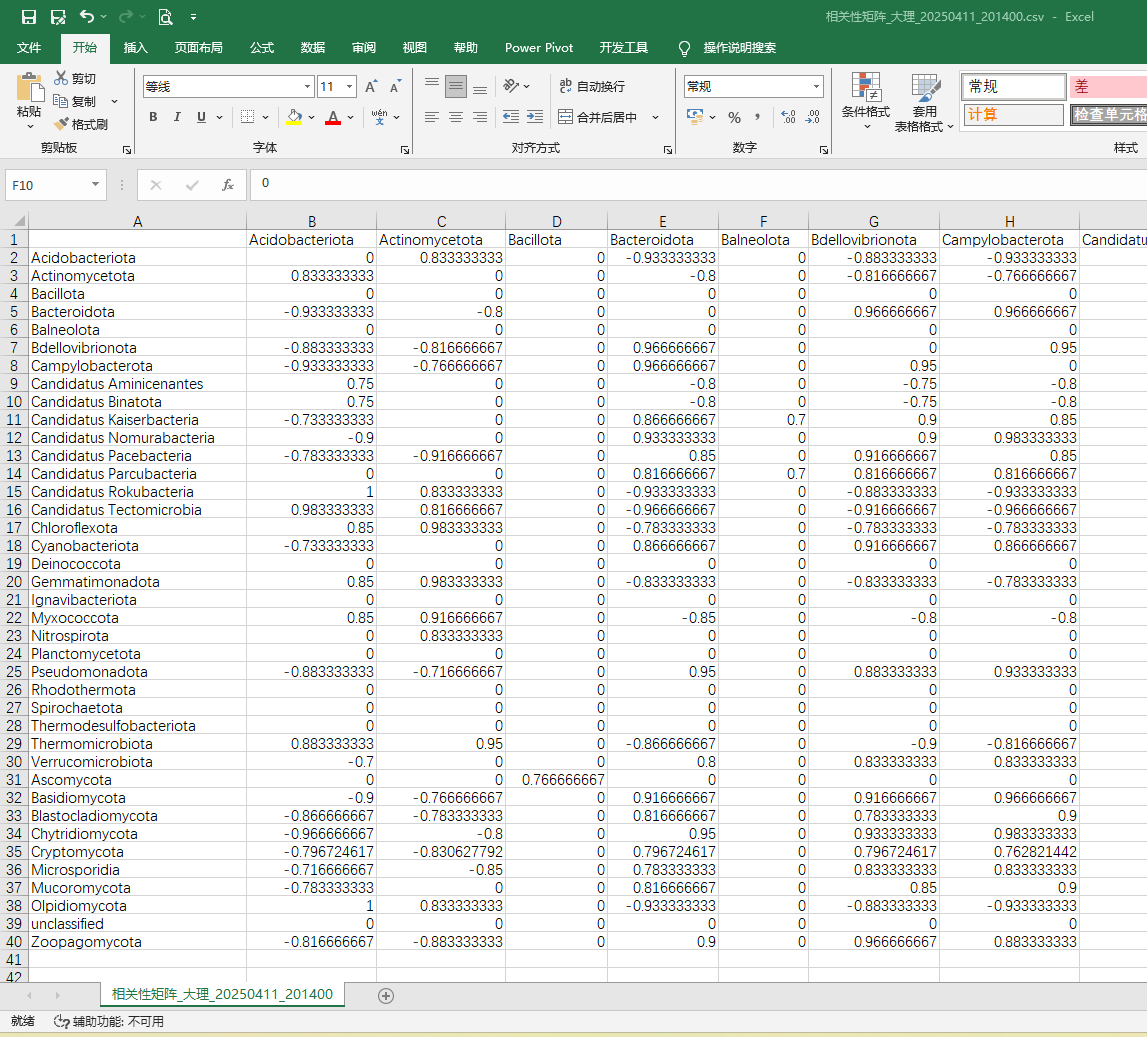
2、节点属性文件
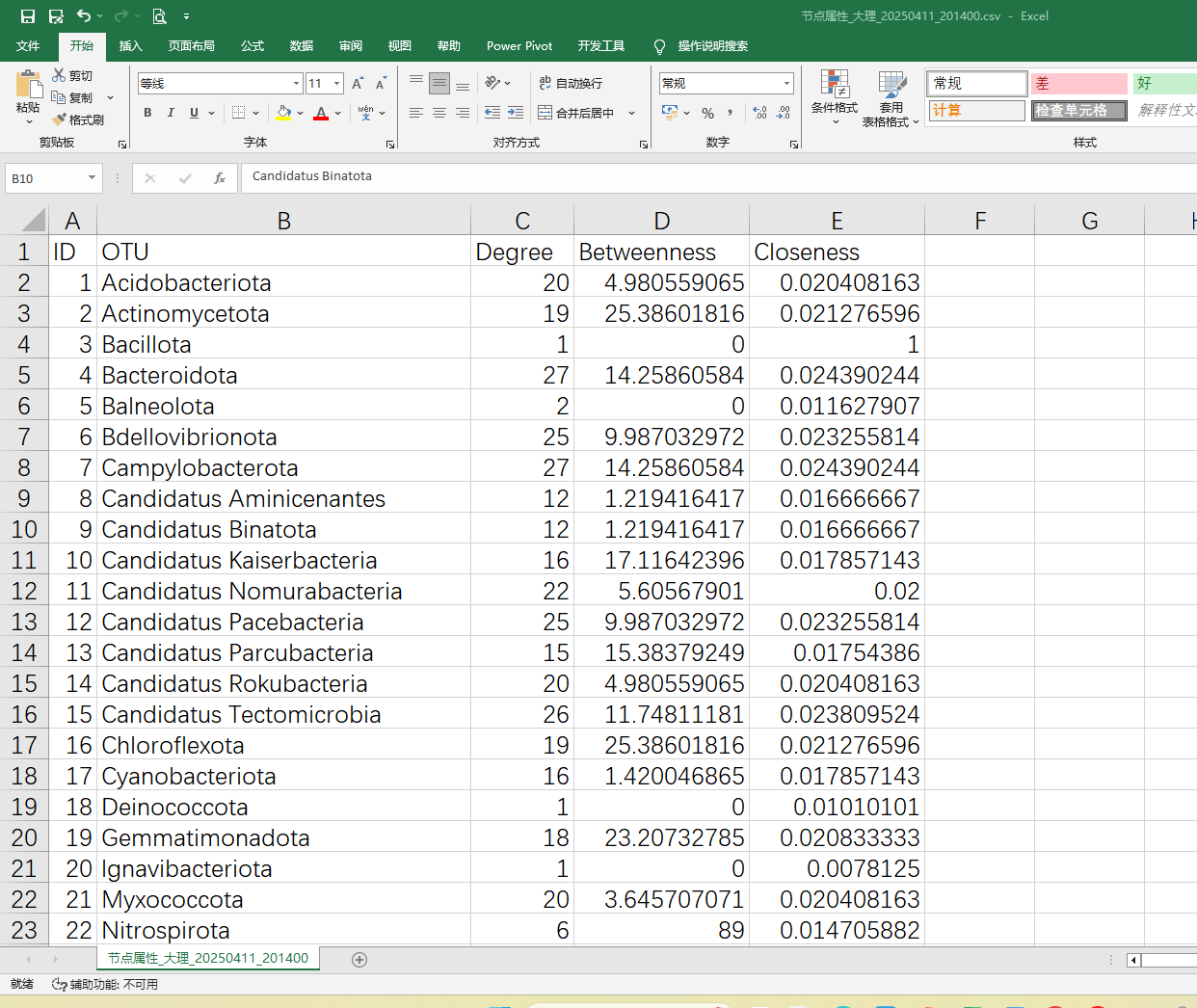
3、网络可视化图
4、分析摘要
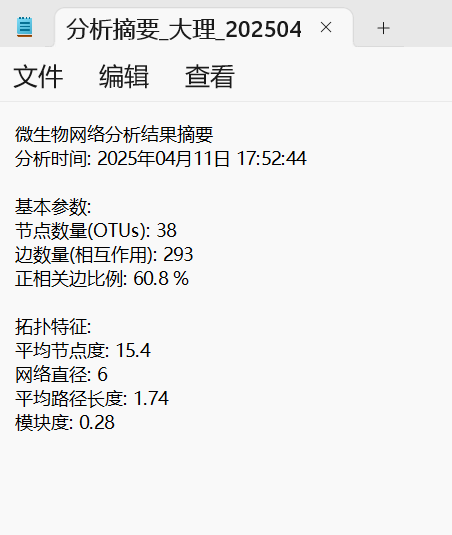
5、边属性文件
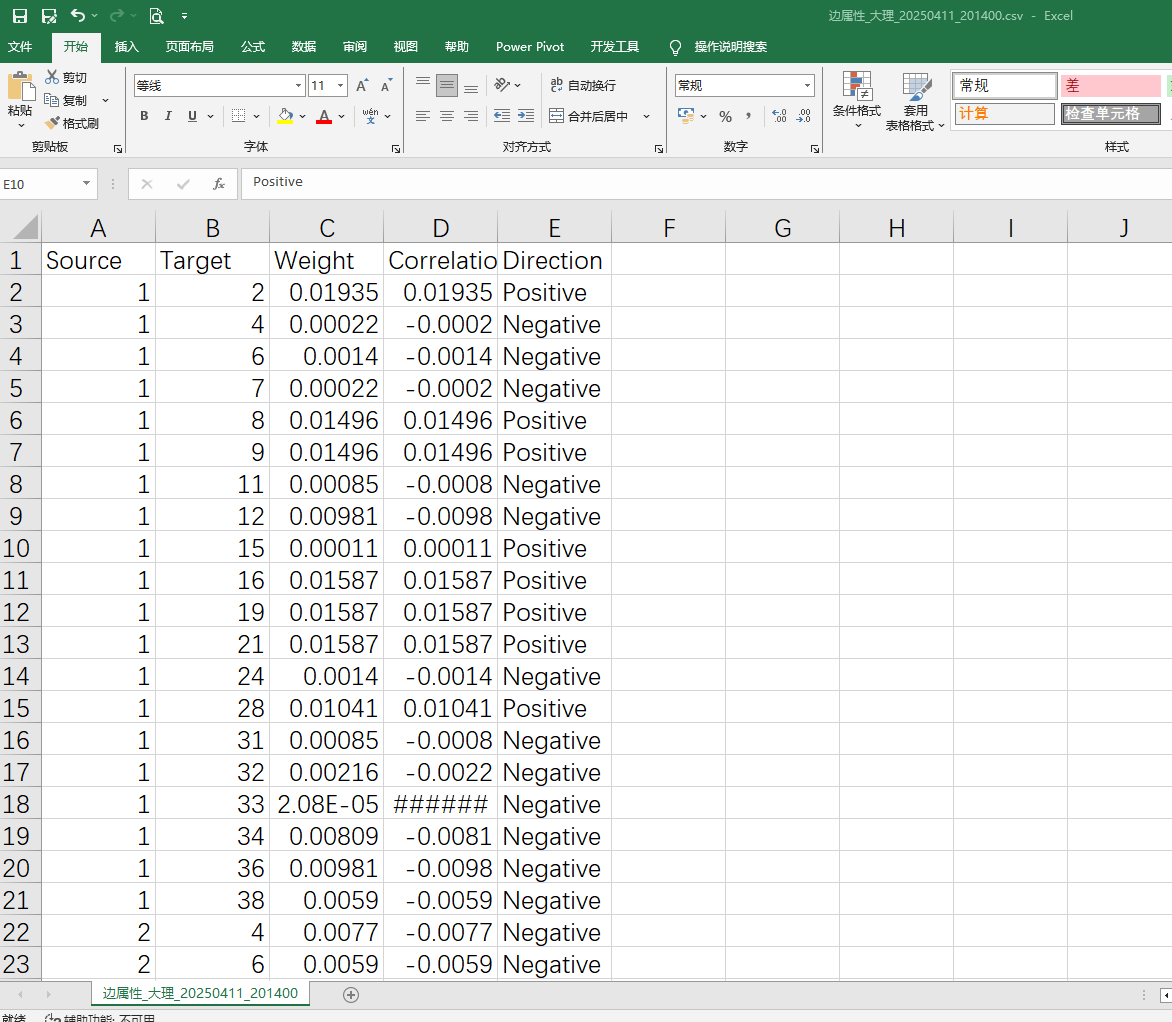
2、Gephi制图
2.1 导入生成的图
2.1.1使用R生成的文件继续修改
因为R语言代码中已经生成了一个【微生物网络_大理_20250411_175031.graphml】文件了,可以直接在gephi中打开。(我试了,不建议使用,主要是它给的文件的id是自定义的n123,没有用我的初始名字不利于后面修改图例,因此建议使用2.1.2的自己导入文件后制图。)
文件------打开-----找到自己的保存文件路径-----点击文件-----打开------选择有向/无向-----确定
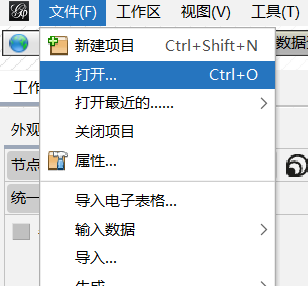
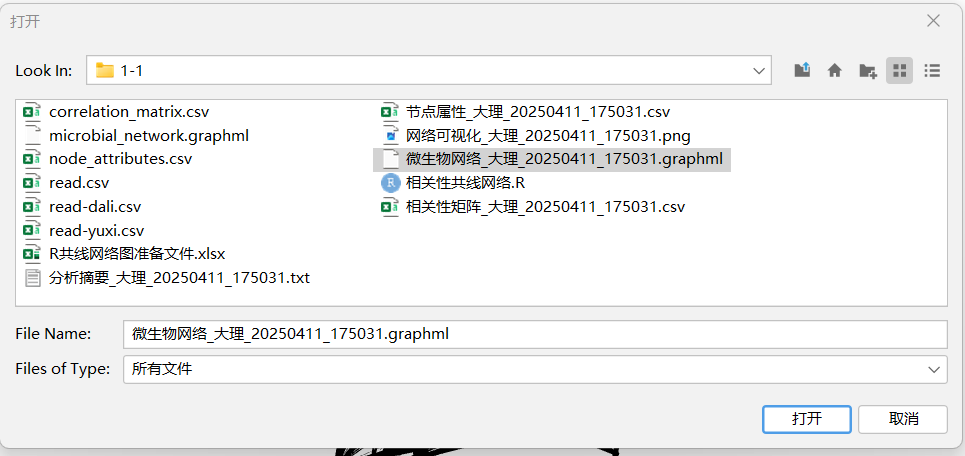
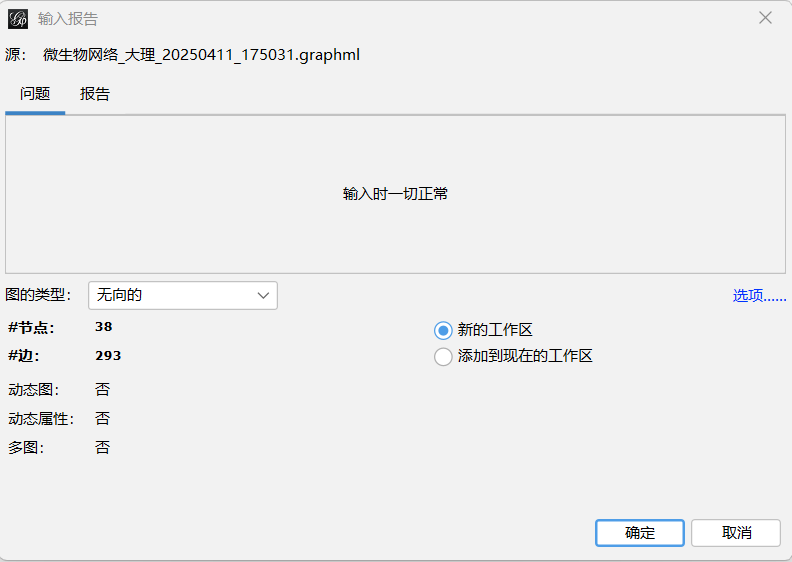
2.1.2 也可以选择自己使用数据表导入
具体方法参考下面的链接或者自己网上找找如何做,下面的是我最开始学gephi的一些链接
输入电子表格------选择节点/边-----导入为:节点/边-------下一步:选择数据类型-------添加到现在的工作区--------确定

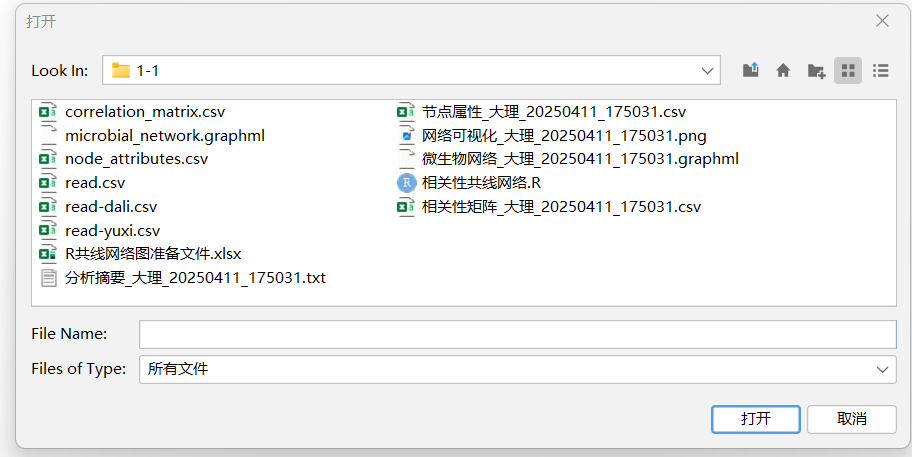
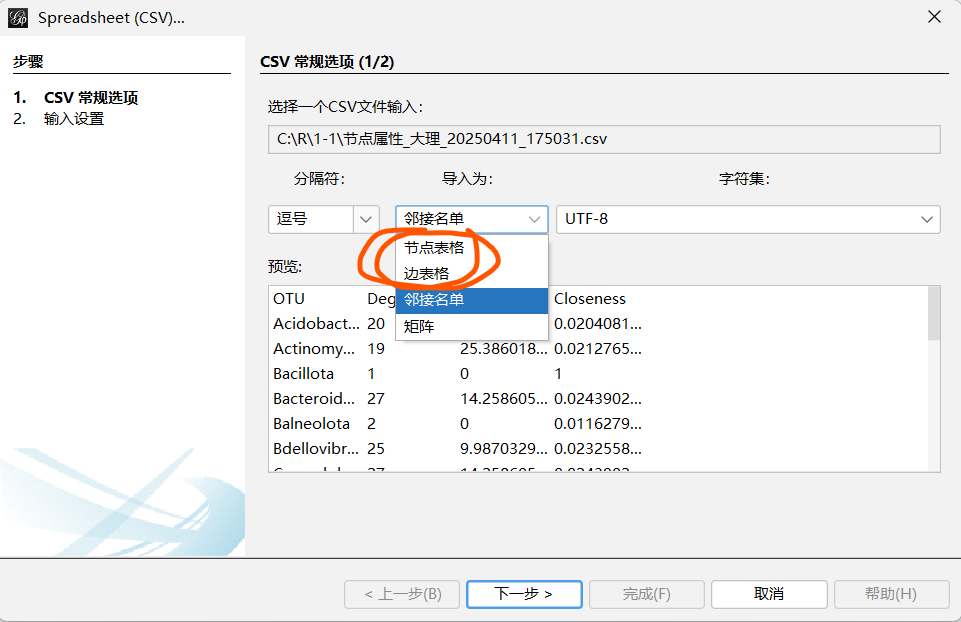
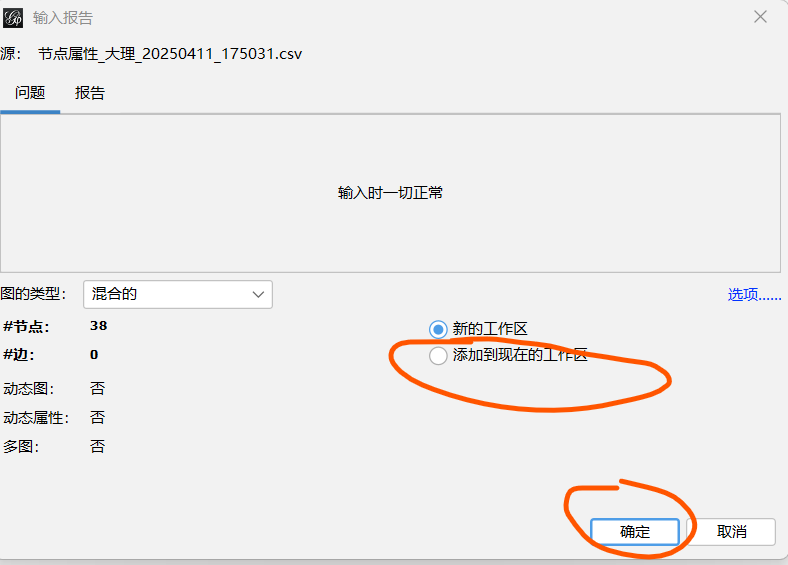
2.2 导入成功后修改样式
2.2.1.这是导入成功的图。
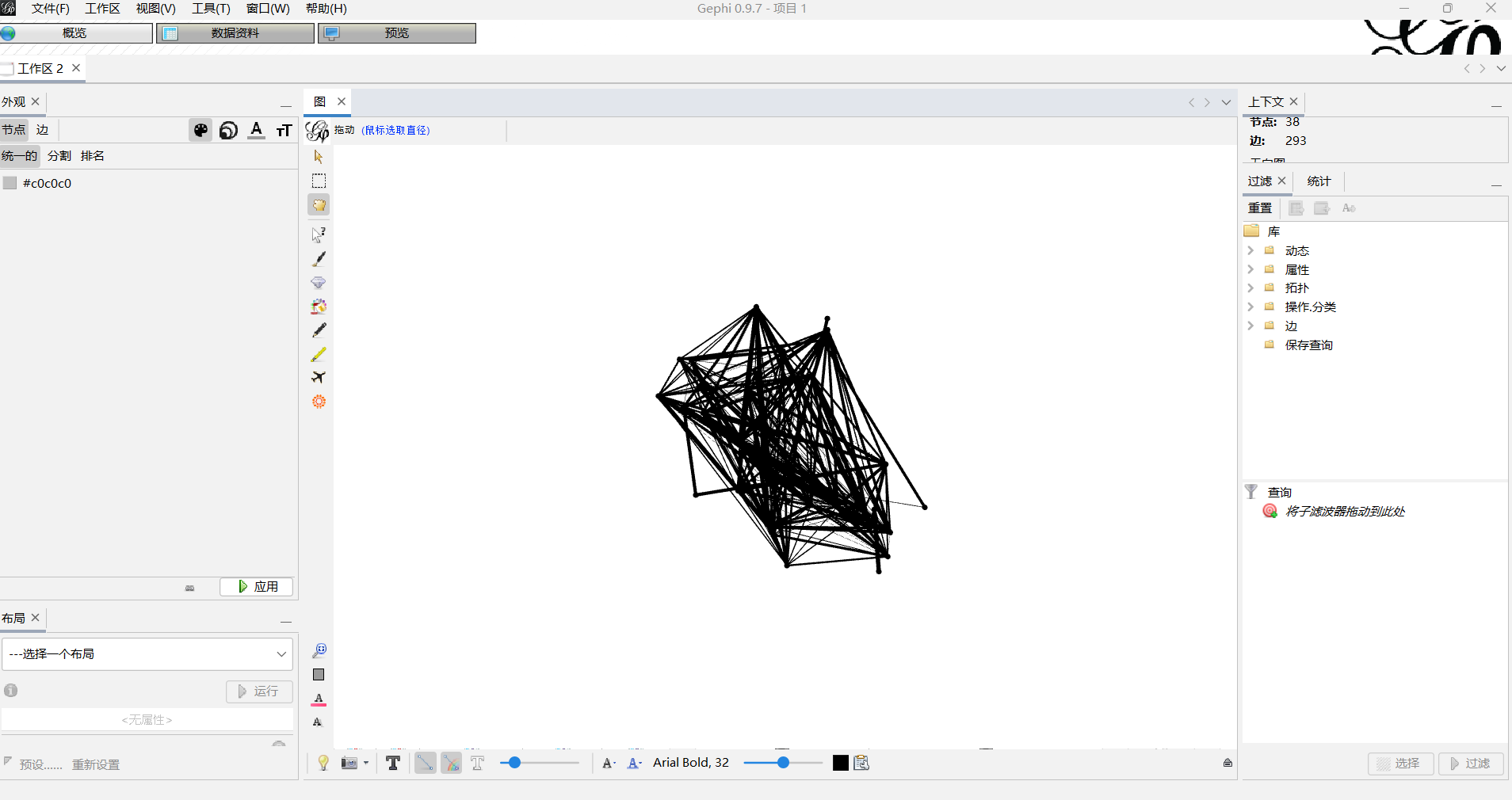
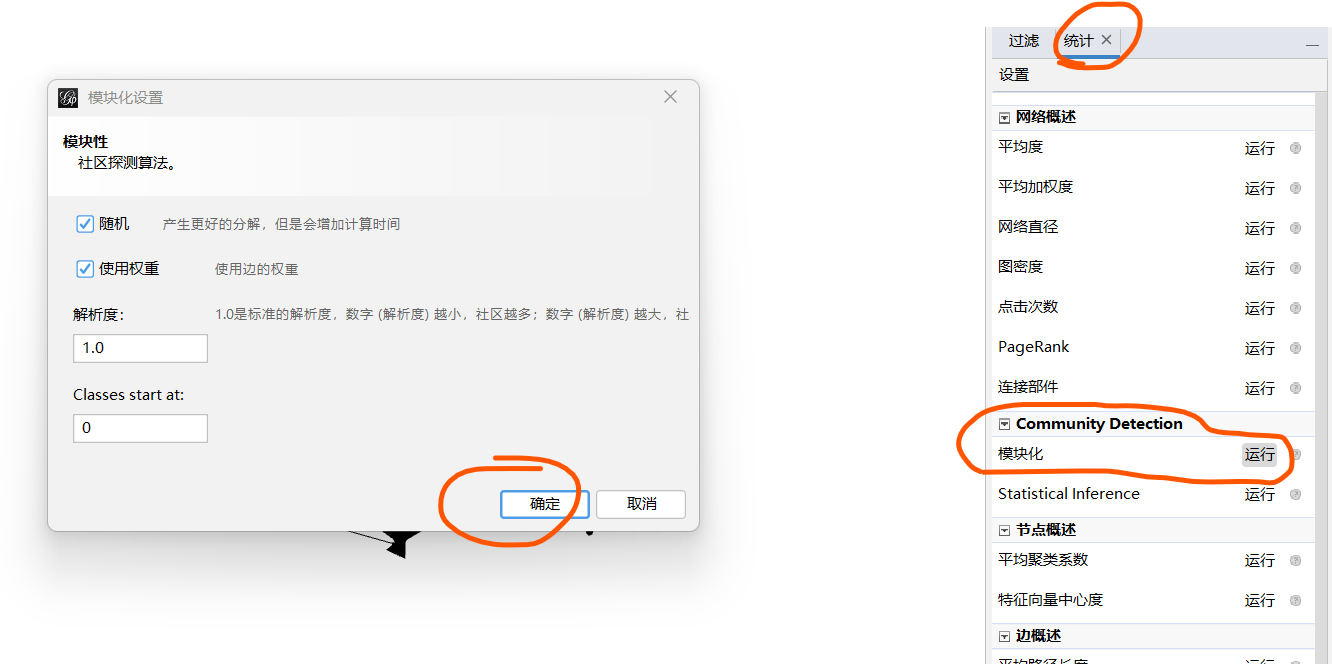
2.2.2 模块化
统计----模块化---确定
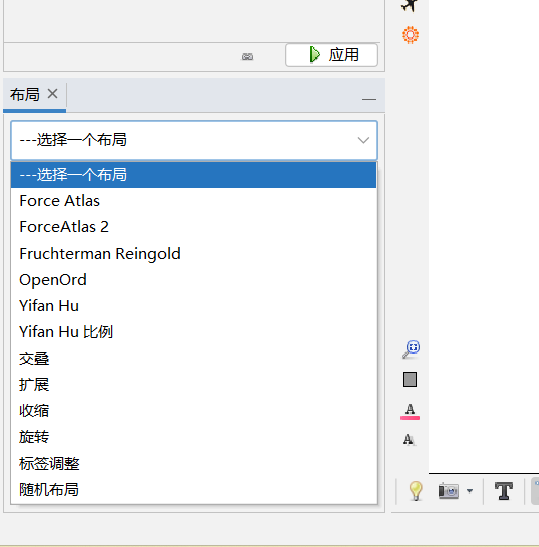
2.2.3 左下角选择布局
布局-----选择一个不局(选一个想要的)-----运行
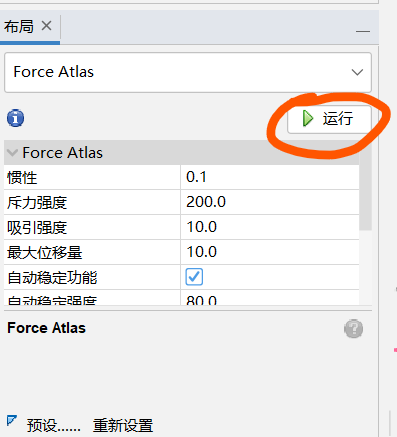
觉得差不多稳定了是自己想要的形状了点击:停止
觉得形状不满意的可以自己填写相关的信息在次运行
2.2.4 修改颜色:
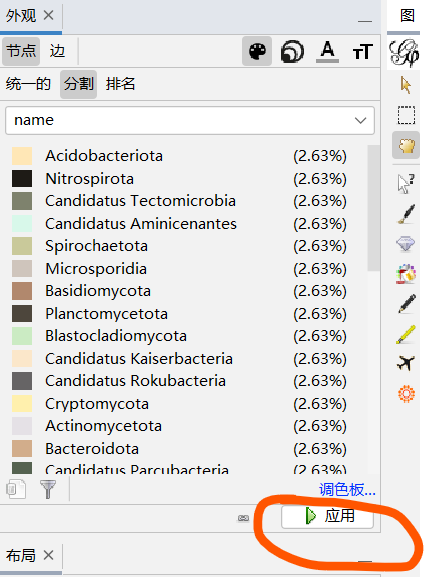
节点------分割----调色盘-------name
但是现在只有前几个有颜色,可以随机生成颜色:
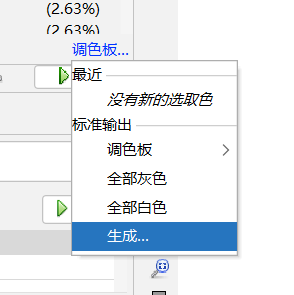
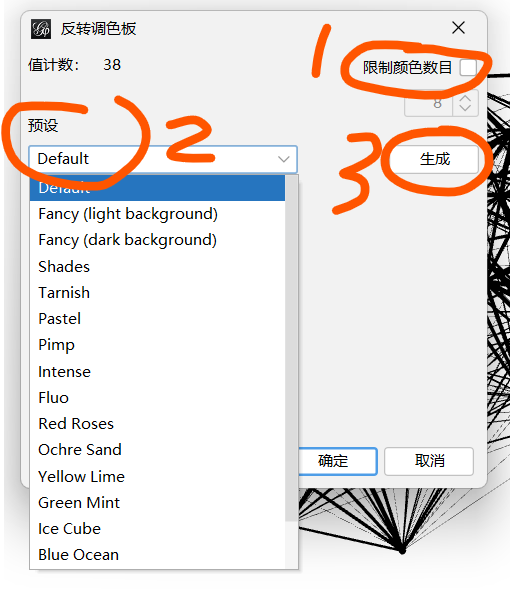
调色板---生成------反转调色板-----取消勾选:限制颜色数目----预设:选择一个喜欢的颜色--------生成------确定-----应用
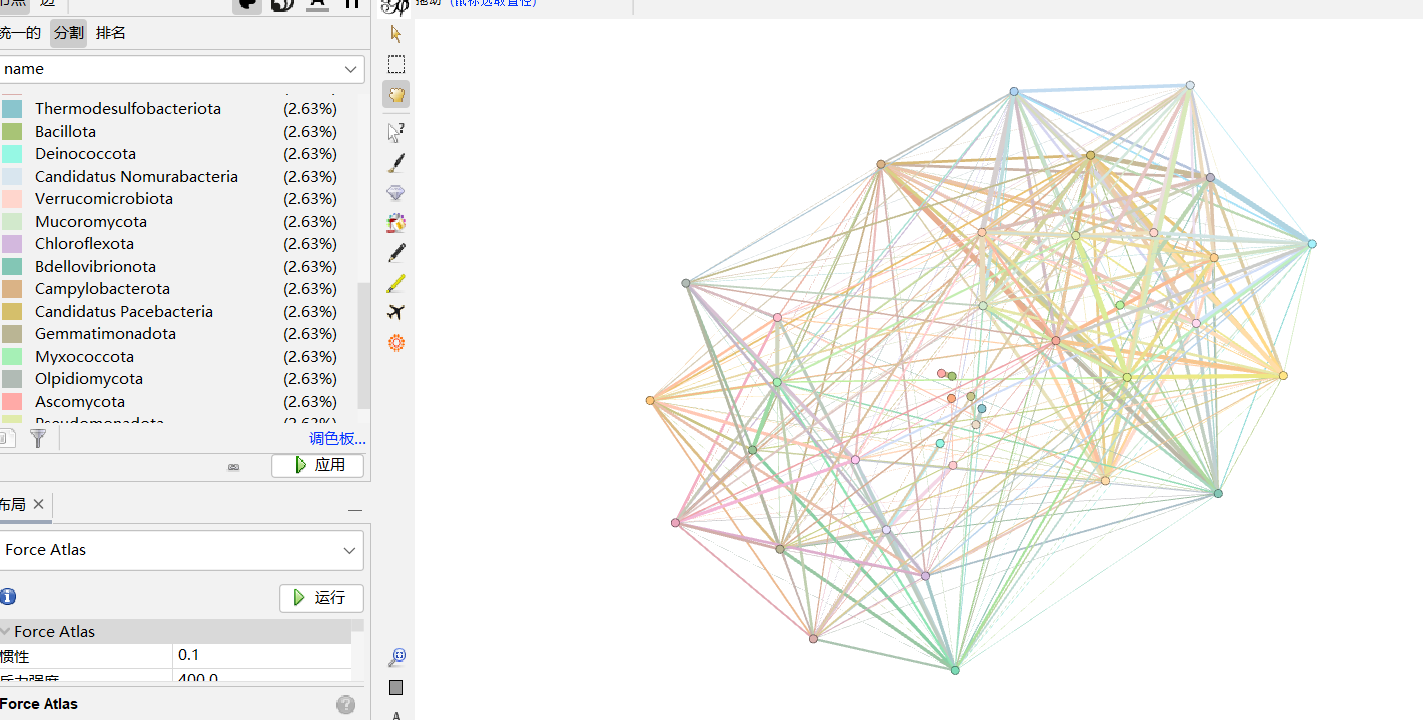
2.2.5 修改线条粗细
2.2.6 修改节点大小
2.2.7 保存gephi
文件-----保存(制图过程中可以多保存几次,因为gephi好像没法撤回操作)
2.3 图片导出
预览---按需设置----刷新-----选择【文件】→【输出】→【SVG/PDF/PNG文件】参考下面的链接
如果没显示图片预览可以点击下面的----预设缩放
2.4 导出到ppt中作图(做图例)
从调整颜色哪里进行截图到ppt中,自行绘制圆圈后吸取截图的对应颜色,制作图例。


3、参考文献成图
D. Gu, X. Xiang, Y. Wu, J. Zeng, X. Lin, Synergy between fungi and bacteria promotes polycyclic aromatic hydrocarbon cometabolism in lignin-amended soil, Journal of Hazardous Materials 425 (2022) 127958. Redirecting.
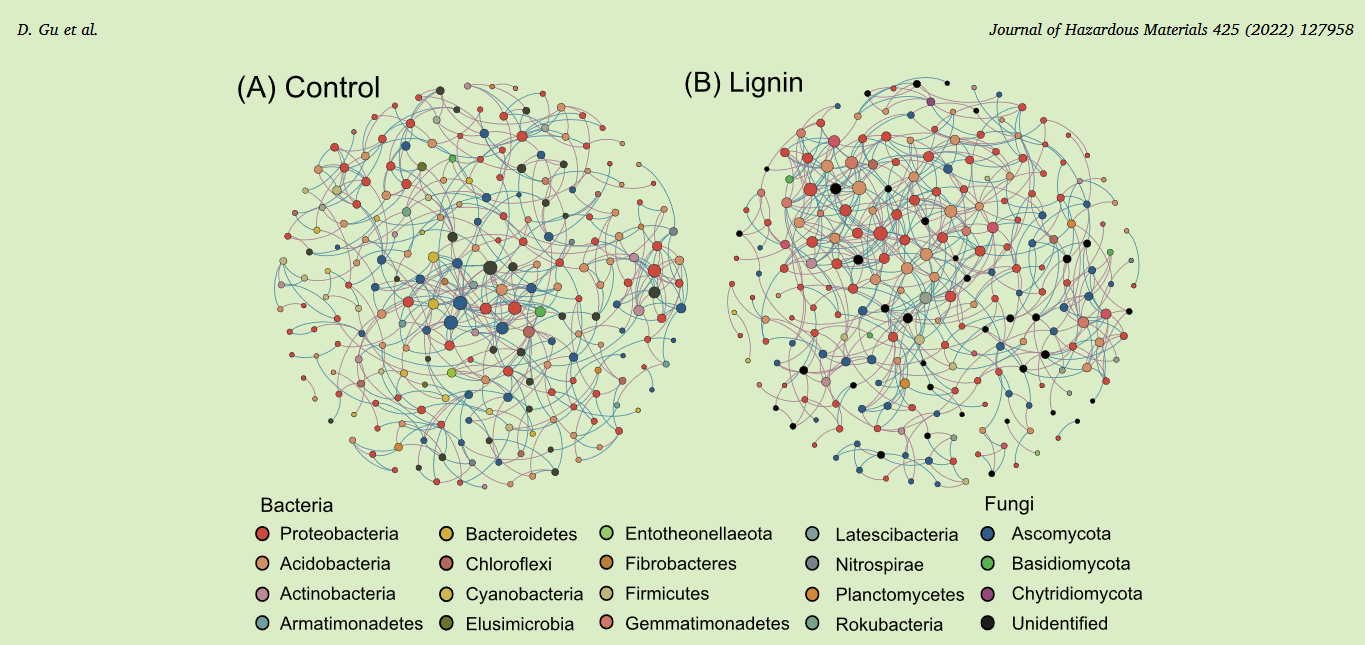
Fig. 6. The co-occurrence networks of (A) the Control treatment and (B) the Lignin treatment. Topological attributes of the network (C) and the interaction relationship in the bacterial-fungal network (D) were calculated.【图6 . ( A ) Control处理和( B ) Lignin处理的共现网络。计算网络的拓扑属性( C )和细菌-真菌网络中的相互作用关系( D )。】
对应的论文分析:
3.5. Co-occurrence networks of bacterial and fungal communities
Co-occurrence networks were built for the Control and Lignin treatments, with nodes representing bacterial and fungal OTUs, and edges being correlation between OTUs based on Spearman rank (Figs. 6A 6B). Network Analyzer tool was used to calculate network topological metrics (Fig. 6C). There were similar node number, characteristic path length and modularity between these two groups when all bacterial and fungal OTUs were considered. However, the edge number, average degree and network density were higher in the Lignin than in the Control, suggesting more links and increased complexity between OTUs with lignin amendment. In particular, lignin increased the number and proportion of both positive and negative interactions between bacterial OTUs, and reduced positive interactions between fungal OTUs (Fig. 6D). Meanwhile, positive links between bacterial and fungal OTUs lignin became abundant, coupled with decreased negative correlations.
3 . 5 .细菌和真菌群落的共现网络
构建Control和Lignin处理的共现网络,节点代表细菌和真菌OTUs,边是基于Spearman rank (图。6A 6B)的OTUs之间的相关性。使用Network Analyzer工具计算网络拓扑指标(图6C )。当考虑所有细菌和真菌OTU时,这两组之间具有相似的节点数、特征路径长度和模块度。然而,Lignin组的边数、平均度和网络密度均高于Control组,表明随着木质素的添加,OTUs之间的联系更多,复杂性增加。特别是,木质素增加了两者之间正负相互作用的数量和比例,减少了真菌OTUs之间的正相互作用(图6D )。同时,细菌和真菌OTUs木质素之间的正联系变得丰富,负相关关系减弱。(有道翻译,不一定准)
预留我的文献位置:等我论文发出来了我就把链接附在这里,欢迎大家转载引用!!!
4、补充:
看到了相关的文章,可以和我这个文章互为补充的,随时补在下面,也欢迎大家评论区补充。

















 1万+
1万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









