




 本文介绍如何使用R软件包affy和simpleaffy进行芯片数据的质量分析,包括读取CEL文件、查看芯片基本信息、显示芯片扫描图像、进行统计分析、MA-plot分析及RNA降解分析等步骤。
本文介绍如何使用R软件包affy和simpleaffy进行芯片数据的质量分析,包括读取CEL文件、查看芯片基本信息、显示芯片扫描图像、进行统计分析、MA-plot分析及RNA降解分析等步骤。
(本文为博客搬家到CSDN后于2014年4月25日重新运行和更新的内容。更换knitr代码加亮模板以示区别。)
TAIR,NASCarray 和 EBI 都有一些公开的免费芯片数据可以下载。本专题使用的数据来自NASCarray(Exp350),也可以用FTP直接下载。下载其中的CEL文件即可(.CEL.gz),下载后解压缩到同一文件夹内。该实验有1个对照和3个处理,各有2个重复,共8张芯片(8个CEL文件)。
为什么要进行芯片质量分析?不是每个人做了实验都会得到高质量的数据,花了钱不一定就有回报,这道理大家都懂。
芯片实验有可能失败,失败的原因可能是技术上的(包括片子本身的质量),也可能是实验设计方面的。芯片质量分析主要检测前者。
1 R软件包安装
使用到两个软件包:affy,simpleaffy:
library(BiocInstaller) biocLite(c("affy", "simpleaffy"))
另外还需要两个辅助软件包:tcltk和scales。tcltk一般R基础安装包都已经装有。
install.packages(c("tcltk", "scales"))
2 读取CEL文件
载入affy软件包:
library(affy) library(tcltk)
选取CEL文件。以下两种方法任选一种即可。
第一种方法是通过选取目录获得某个目录内(包括子目录)的所有cel文件:
# 用choose.dir函数选择文件夹 dir <- tk_choose.dir(caption = "Select folder") # 列出CEL文件,保存到变量 cel.files <- list.files(path = dir, pattern = ".+\\.cel$", ignore.case = TRUE, full.names = TRUE, recursive = TRUE) # 查看文件名 basename(cel.files)
第二种方法是通过文件选取选择目录内部分或全部cel文件:
# 建立文件过滤器 filters <- matrix(c("CEL file", ".[Cc][Ee][Ll]", "All", ".*"), ncol = 2, byrow = T) # 使用tk_choose.files函数选择文件 cel.files <- tk_choose.files(caption = "Select CELs", multi = TRUE, filters = filters, index = 1) # 注意:较老版本的tk函数有bug,列表的第一个文件名可能是错的 basename(cel.files)
## [1] "NRID9780_Zarka_2-1_MT-0HCA(SOIL)_Rep1_ATH1.CEL" ## [2] "NRID9781_Zarka_2-2_MT-0HCB(SOIL)_Rep2_ATH1.CEL" ## [3] "NRID9782_Zarka_2-3_MT-1HCA(SOIL)_Rep1_ATH1.CEL" ## [4] "NRID9783_Zarka_2-4_MT-1HCB(SOIL)_Rep2_ATH1.CEL" ## [5] "NRID9784_Zarka_2-5_MT-24HCA(SOIL)_Rep1_ATH1.CEL" ## [6] "NRID9785_Zarka_2-6_MT-24HCB(SOIL)_Rep2_ATH1.CEL" ## [7] "NRID9786_Zarka_2-7_MT-7DCA(SOIL)_Rep1_ATH1.CEL" ## [8] "NRID9787_Zarka_2-8_MT-7DCB(SOIL)_Rep2_ATH1.CEL"
读取CEL文件数据使用ReadAffy函数,它的参数为:
# Not run. 函数说明,请不要运行下面代码 ReadAffy(..., filenames = character(0), widget = getOption("BioC")$affy$use.widgets, compress = getOption("BioC")$affy$compress.cel, celfile.path = NULL, sampleNames = NULL, phenoData = NULL, description = NULL, notes = "", rm.mask = FALSE, rm.outliers = FALSE, rm.extra = FALSE, verbose = FALSE, sd = FALSE, cdfname = NULL)
除文件名外我们使用函数的默认参数读取CEL文件:
data.raw <- ReadAffy(filenames = cel.files)
读入芯片的默认样品名称是文件名,用sampleNames函数查看或修改:
sampleNames(data.raw)
## [1] "NRID9780_Zarka_2-1_MT-0HCA(SOIL)_Rep1_ATH1.CEL" ## [2] "NRID9781_Zarka_2-2_MT-0HCB(SOIL)_Rep2_ATH1.CEL" ## [3] "NRID9782_Zarka_2-3_MT-1HCA(SOIL)_Rep1_ATH1.CEL" ## [4] "NRID9783_Zarka_2-4_MT-1HCB(SOIL)_Rep2_ATH1.CEL" ## [5] "NRID9784_Zarka_2-5_MT-24HCA(SOIL)_Rep1_ATH1.CEL" ## [6] "NRID9785_Zarka_2-6_MT-24HCB(SOIL)_Rep2_ATH1.CEL" ## [7] "NRID9786_Zarka_2-7_MT-7DCA(SOIL)_Rep1_ATH1.CEL" ## [8] "NRID9787_Zarka_2-8_MT-7DCB(SOIL)_Rep2_ATH1.CEL"
sampleNames(data.raw) <- paste("CHIP", 1:length(cel.files), sep = "-") sampleNames(data.raw)
## [1] "CHIP-1" "CHIP-2" "CHIP-3" "CHIP-4" "CHIP-5" "CHIP-6" "CHIP-7" "CHIP-8"
3 查看芯片的基本信息
Phenotypic data数据可能有用,可以修改成你需要的内容,用pData函数查看和修改:
pData(data.raw)
## sample ## CHIP-1 1 ## CHIP-2 2 ## CHIP-3 3 ## CHIP-4 4 ## CHIP-5 5 ## CHIP-6 6 ## CHIP-7 7 ## CHIP-8 8
pData(data.raw)$Treatment <- gl(2, 1, length = length(cel.files), labels = c("CK", "T")) pData(data.raw)
## sample Treatment ## CHIP-1 1 CK ## CHIP-2 2 T ## CHIP-3 3 CK ## CHIP-4 4 T ## CHIP-5 5 CK ## CHIP-6 6 T ## CHIP-7 7 CK ## CHIP-8 8 T
PM和MM查看:
# Perfect-match probes pm.data <- pm(data.raw) head(pm.data)
## CHIP-1 CHIP-2 CHIP-3 CHIP-4 CHIP-5 CHIP-6 CHIP-7 CHIP-8 ## 501131 127.0 166.3 112.0 139.8 111.3 85.5 126.3 102.8 ## 251604 118.5 105.0 82.0 101.5 94.0 81.3 103.8 103.0 ## 261891 117.0 90.5 113.0 101.8 99.3 107.0 85.3 85.3 ## 230387 140.5 113.5 94.8 137.5 117.3 112.5 124.3 114.0 ## 217334 227.3 192.5 174.0 192.8 162.3 163.3 235.0 195.8 ## 451116 135.0 122.0 86.8 93.3 83.8 87.3 97.3 83.5
# Mis-match probes mm.data <- mm(data.raw) head(mm.data)
## CHIP-1 CHIP-2 CHIP-3 CHIP-4 CHIP-5 CHIP-6 CHIP-7 CHIP-8 ## 501843 89.0 88.0 80.5 91.0 77.0 75.0 79.0 72.0 ## 252316 134.3 77.3 77.0 107.8 98.5 75.0 99.5 71.3 ## 262603 119.3 90.5 82.0 86.3 93.0 89.3 94.5 83.8 ## 231099 123.5 94.5 76.5 95.0 89.3 87.8 95.5 91.5 ## 218046 110.3 93.0 74.8 100.5 86.0 89.5 104.5 102.3 ## 451828 127.5 77.0 80.3 94.5 72.3 79.0 86.3 67.8
4 显示芯片扫描图像(灰度)
# 芯片数量 n.cel <- length(cel.files) par(mfrow = c(ceiling(n.cel/2), 2)) par(mar = c(0.5, 0.5, 2, 0.5)) # 设置调色板颜色为灰度 pallette.gray <- c(rep(gray(0:10/10), times = seq(1, 41, by = 4))) # 通过for循环逐个作图 for (i in 1:n.cel) image(data.raw[, i], col = pallette.gray)

如果芯片图像有斑块现象就很可能是坏片。
5 对灰度值做简单统计分析
5.1 箱线图
par(mfrow = c(1, 1)) par(mar = c(4, 4, 3, 0.5)) par(cex = 0.7) if (n.cel > 40) par(cex = 0.5) # rainbow是R的一个函数,用于产生彩虹色 cols <- rainbow(n.cel * 1.2) boxplot(data.raw, col = cols, xlab = "Sample", ylab = "Log intensity")
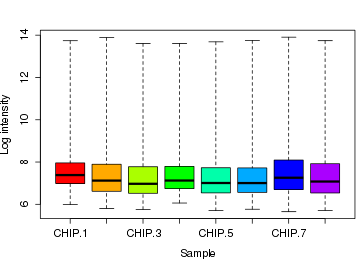
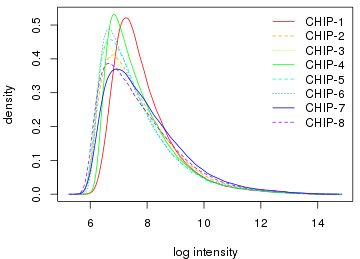
5.2 histogram曲线
par(mar = c(4, 4, 0.5, 0.5)) hist(data.raw, lty = 1:3, col = cols) legend("topright", legend = sampleNames(data.raw), lty = 1:3, col = cols, box.col = "transparent", xpd = TRUE)
6 MA-plot分析
par(mfrow = c(ceiling(n.cel/2), 2)) par(mar = c(3, 3, 2, 0.5)) par(tcl = 0.2) par(mgp = c(2, 0.5, 0)) require(scales) col <- alpha("seagreen", alpha = 0.01) MAplot(data.raw, col = col, loess.col = "red", cex = 0.9)
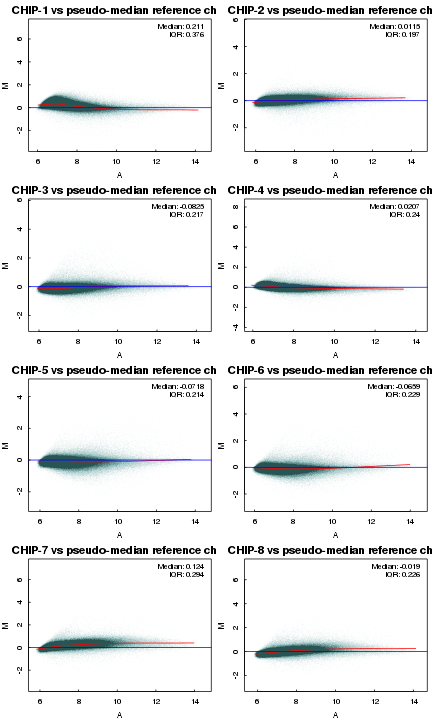
IQR差别大的芯片可能有问题,但芯片能不能用得看具体情况(参考其他指标)而定。
7 RNA降解分析
par(mfrow = c(1, 1)) par(mar = c(4, 4, 3, 0.5)) RNAdeg <- AffyRNAdeg(data.raw) summaryAffyRNAdeg(RNAdeg)
## CHIP-1 CHIP-2 CHIP-3 CHIP-4 CHIP-5 CHIP-6 CHIP-7 ## slope 1.71e+00 1.67e+00 1.82e+00 1.9e+00 1.60e+00 1.73e+00 1.72e+00 ## pvalue 2.31e-10 5.11e-11 4.74e-11 3.0e-11 2.84e-11 3.39e-12 1.31e-10 ## CHIP-8 ## slope 1.61e+00 ## pvalue 7.84e-11
plotAffyRNAdeg(RNAdeg, cols = cols) legend("topleft", legend = sampleNames(data.raw), lty = 1, col = cols, box.col = "transparent", xpd = TRUE) box()
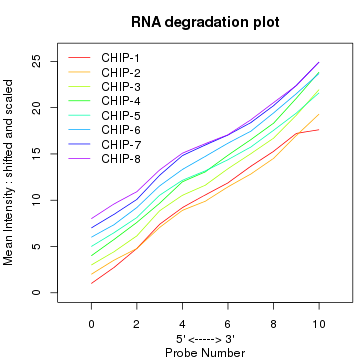
理想状况下各样品的线(分段)是平行的。从上面图上看芯片1可能有点问题。
8 用simpleaffy包进行分析
library(simpleaffy) # 计算芯片质量数据 qc.data <- qc(data.raw) # 平均背景值,如果太大则表示可能有问题 (avbg.data <- as.data.frame(sort(avbg(qc.data))))
## sort(avbg(qc.data)) ## CHIP-8 60.74 ## CHIP-5 63.53 ## CHIP-2 63.71 ## CHIP-3 63.92 ## CHIP-7 63.92 ## CHIP-6 66.59 ## CHIP-1 78.95 ## CHIP-4 79.61
# 样品的scale factor (sfs.data <- sort(sfs(qc.data)))
## [1] 0.5689 0.6235 0.6905 0.6920 0.7660 0.8179 0.8191 0.8386
# affy建议每个样品间的sf差异不能超过3倍 max(sfs.data)/min(sfs.data)
## [1] 1.474
# 表达基因所占的比例,太小则表示有问题 as.data.frame(percent.present(qc.data))
## percent.present(qc.data) ## CHIP-1.present 58.27 ## CHIP-2.present 62.10 ## CHIP-3.present 62.98 ## CHIP-4.present 60.95 ## CHIP-5.present 58.02 ## CHIP-6.present 59.35 ## CHIP-7.present 62.66 ## CHIP-8.present 62.30
# 内参基因的表达比例 ratios(qc.data)
## actin3/actin5 actin3/actinM gapdh3/gapdh5 gapdh3/gapdhM ## CHIP-1 0.3860 -0.297736 0.3118 -0.9426 ## CHIP-2 0.3999 -0.179446 0.3333 -0.6741 ## CHIP-3 0.3891 -0.005161 0.5414 -0.7286 ## CHIP-4 0.4889 -0.152291 0.5449 -0.7081 ## CHIP-5 0.2049 -0.348223 0.4260 -0.6383 ## CHIP-6 0.4554 -0.039076 0.2426 -0.8057 ## CHIP-7 0.5528 -0.226408 0.4426 -0.5121 ## CHIP-8 0.4545 -0.152246 0.2308 -0.8548
9 代码的运行环境(Session Info)
sessionInfo()
## R version 3.1.0 (2014-04-10) ## Platform: x86_64-pc-linux-gnu (64-bit) ## ## locale: ## [1] LC_CTYPE=zh_CN.UTF-8 LC_NUMERIC=C ## [3] LC_TIME=zh_CN.UTF-8 LC_COLLATE=zh_CN.UTF-8 ## [5] LC_MONETARY=zh_CN.UTF-8 LC_MESSAGES=zh_CN.UTF-8 ## [7] LC_PAPER=zh_CN.UTF-8 LC_NAME=C ## [9] LC_ADDRESS=C LC_TELEPHONE=C ## [11] LC_MEASUREMENT=zh_CN.UTF-8 LC_IDENTIFICATION=C ## ## attached base packages: ## [1] parallel tcltk stats graphics grDevices utils datasets ## [8] methods base ## ## other attached packages: ## [1] simpleaffy_2.41.0 gcrma_2.37.0 genefilter_1.47.0 ## [4] scales_0.2.4 ath1121501cdf_2.14.0 affy_1.43.0 ## [7] Biobase_2.25.0 BiocGenerics_0.11.0 zblog_0.1.0 ## [10] knitr_1.5 ## ## loaded via a namespace (and not attached): ## [1] affyio_1.33.0 annotate_1.43.2 AnnotationDbi_1.27.3 ## [4] BiocInstaller_1.15.2 Biostrings_2.33.3 colorspace_1.2-4 ## [7] DBI_0.2-7 evaluate_0.5.3 formatR_0.10 ## [10] GenomeInfoDb_1.1.2 highr_0.3 IRanges_1.99.2 ## [13] munsell_0.4.2 plyr_1.8.1 preprocessCore_1.27.0 ## [16] Rcpp_0.11.1 RSQLite_0.11.4 S4Vectors_0.0.2 ## [19] splines_3.1.0 stats4_3.1.0 stringr_0.6.2 ## [22] survival_2.37-7 tools_3.1.0 XML_3.98-1.1 ## [25] xtable_1.7-3 XVector_0.5.3 zlibbioc_1.11.1

















 4716
4716

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









