




工具链接:https://www.henbio.com/tools
转录组测序(RNA-seq)是生命科学研究中一项广泛应用的技术,它在众多生命科学领域都发挥了重要作用。转录组学研究能够为基因组学提供关键的功能信息补充,并且为揭示生物学系统的基本原理打开了一扇窗口。
为了帮助那些没有任何编程基础的用户轻松进行转录组数据分析,HiOmics云平台推出了转录组一键式分析工具。用户可以利用二代双末端原始测序数据(fastq文件),通过该工具完成质量控制、样本相关性分析、主成分分析(PCA)、基因表达量测定、基因表达差异分析以及转录组KEGG/GO/GSEA富集分析等一系列流程。用户只需在网页上进行简单的鼠标点击操作,经过5个步骤,就能以最高效的方式、最低的成本完成转录组数据分析。
工具链接:https://www.henbio.com/tools
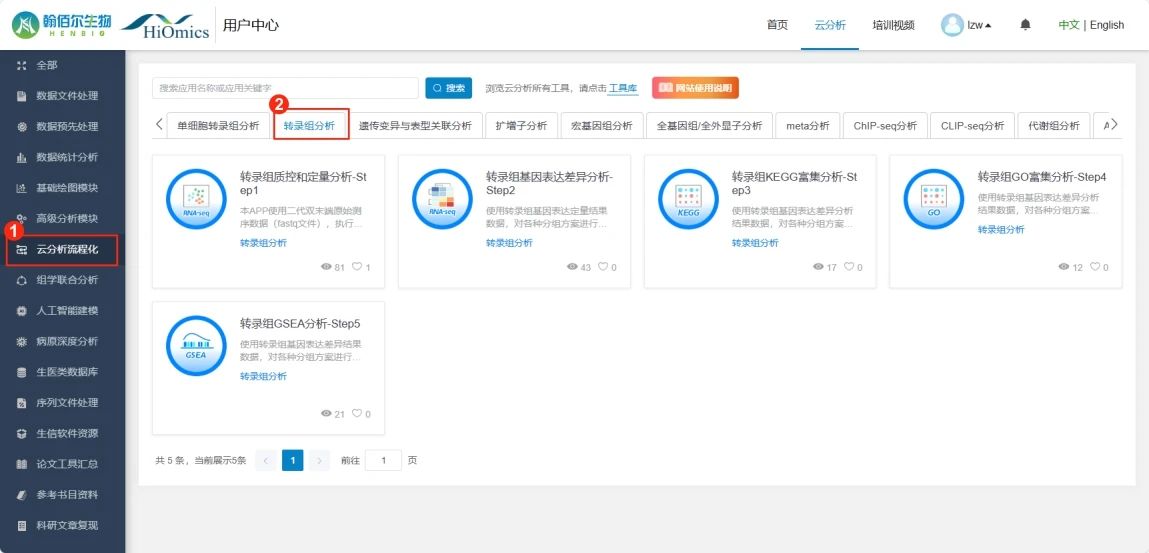
进入【云分析流程化】→【转录组分析】
第一步:转录组质控和定量分析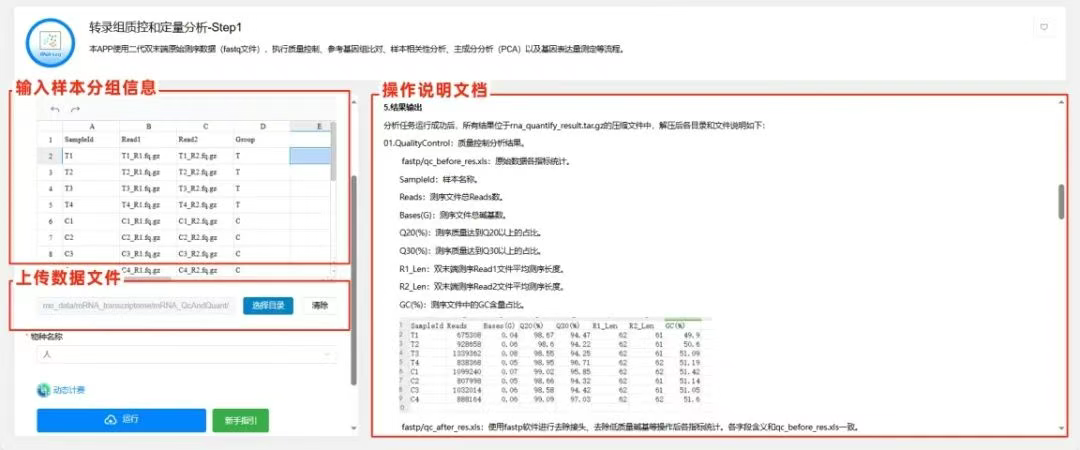
数据准备
按照以下格式输入包含样本和数据文件信息的表格,共有四列:
1)第一列:样本名称(SampleId)。
2)第二列:双端测序的第一个文件名称(Read1),文件通常以R1.fq.gz或1.fastq.gz等结尾。
3)第三列:双端测序的第二个文件名(Read2),文件通常以R2.fq.gz或2.fastq.gz等结尾。
4)第四列:样本分组名称(Group)。
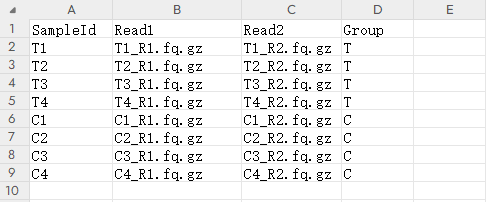
用户可以先在Excel中整理这些信息,然后使用复制(Ctrl+C)和粘贴(Ctrl+V)操作将数据输入到网页的表格输入框中。
数据上传
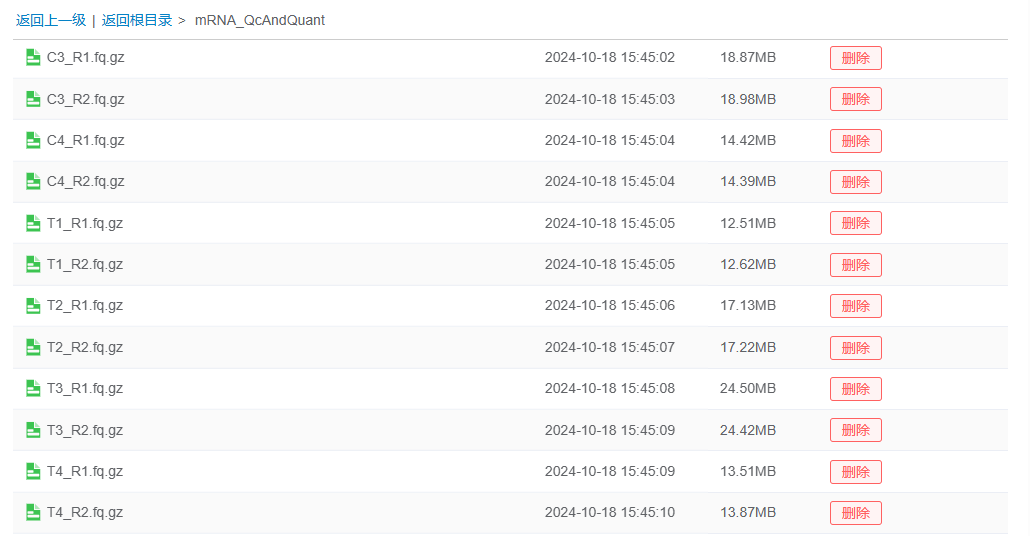
数据文件所在目录:选择样本描述文件中的Read1、Read2对应的测序文件所在的目录。在分析前,应先把需要分析的数据文件上传到平台特定目录,确保所有需要一起分析的文件位于同一个目录里面。

选择样本物种→点击【运行】,服务器运算后输出hisat2比对+featureCounts定量流程等结果文件,其中的rna_quantify_result.tar.gz文件要用作为下一步分析的输入数据文件。
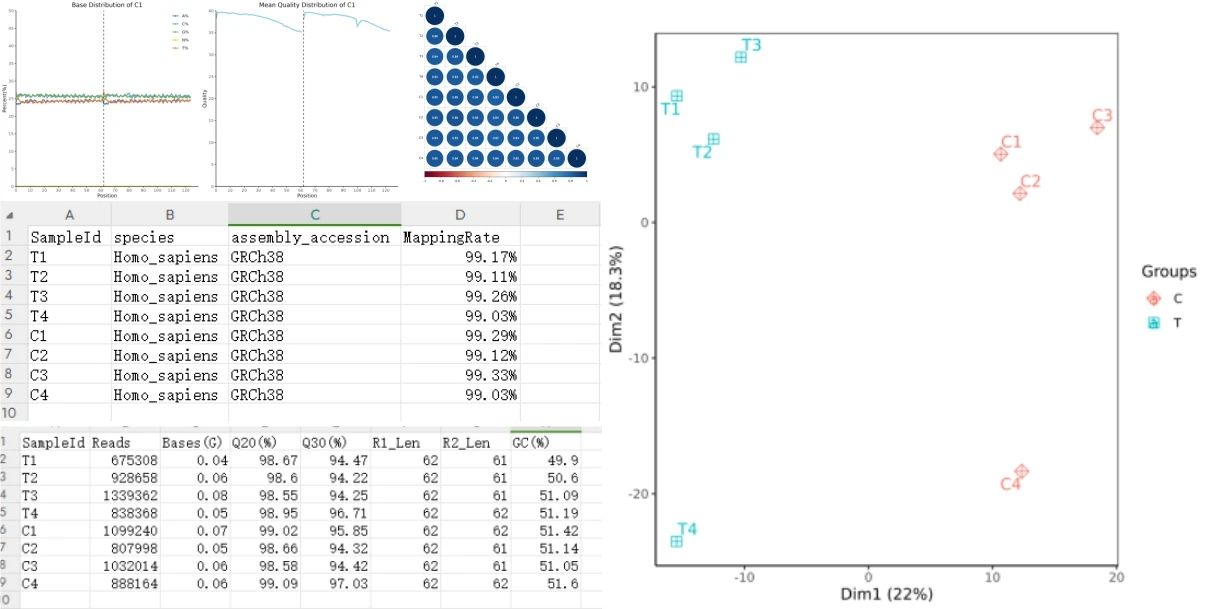
输出结果为质量控制、参考基因组比对、样本相关性分析、主成分分析(PCA)以及基因表达量测定、基因表达矩阵。
第二步:转录组基因表达差异分析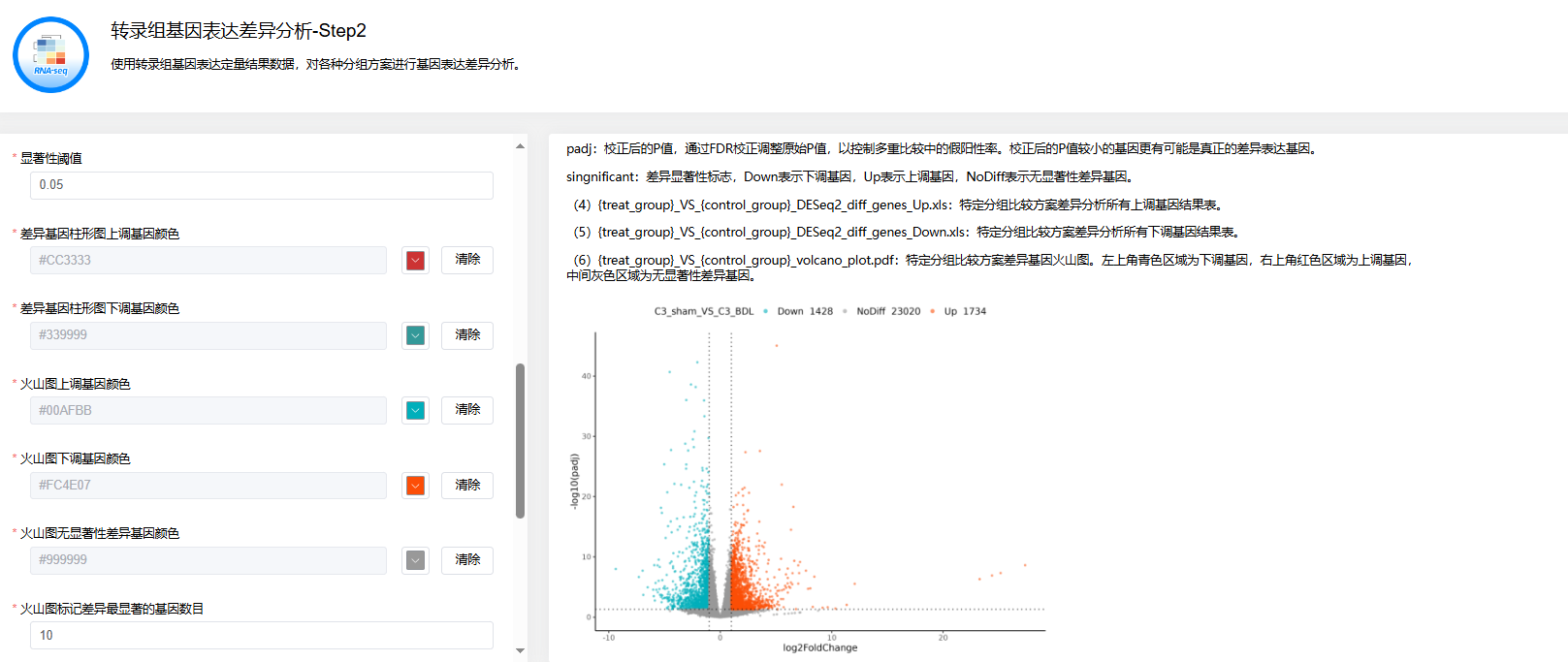
点击【转录组基因表达差异分析】→选择上一步【转录组质控和定量分析】后得到的rna_quantify_result.tar.gz文件→设置【样本分组比较方案】【差异倍数】【差异显著性统计方法】【显著性阈值】等参数→点击【运行】
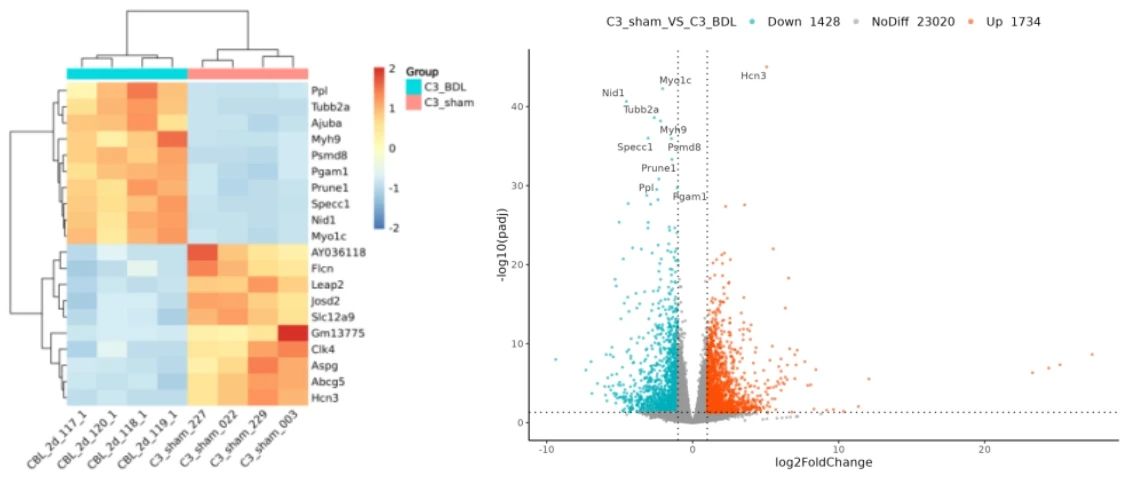
输出结果为差异基因绘制的热图、火山图,以及名为04.DiffGenesAnalysis.tar.gz压缩包文件,作为下一步富集分析的输入数据文件。
第三步:转录组KEGG富集分析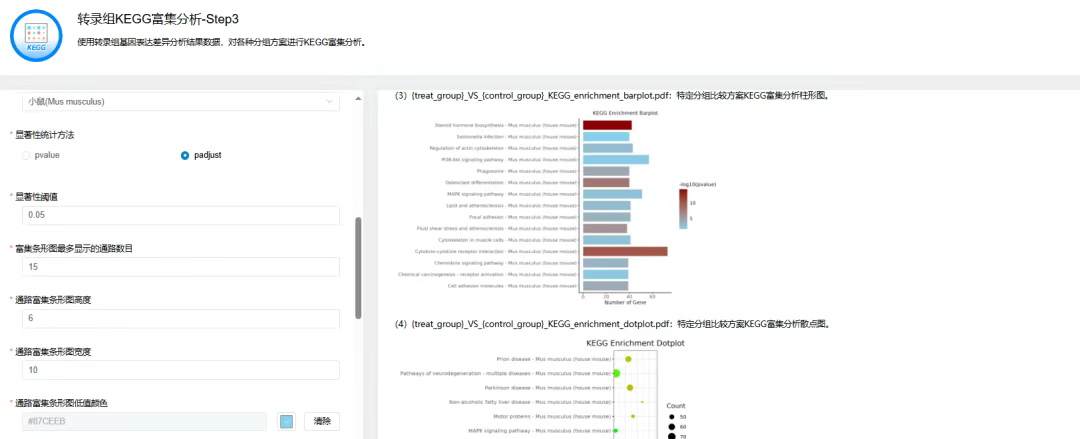
点击【转录组KEGG富集分析】→选择上一步【转录组基因表达差异分析】后得到的04.DiffGenesAnalysis.tar.gz文件→设置【样本分组比较方案】【物种名称】【显著性统计方法】【差异显著性统计方法】【显著性阈值】等参数→点击【运行】

输出结果为KEGG富集分析结果表、柱形图、散点图、通路示意图
第四步:转录组GO富集分析
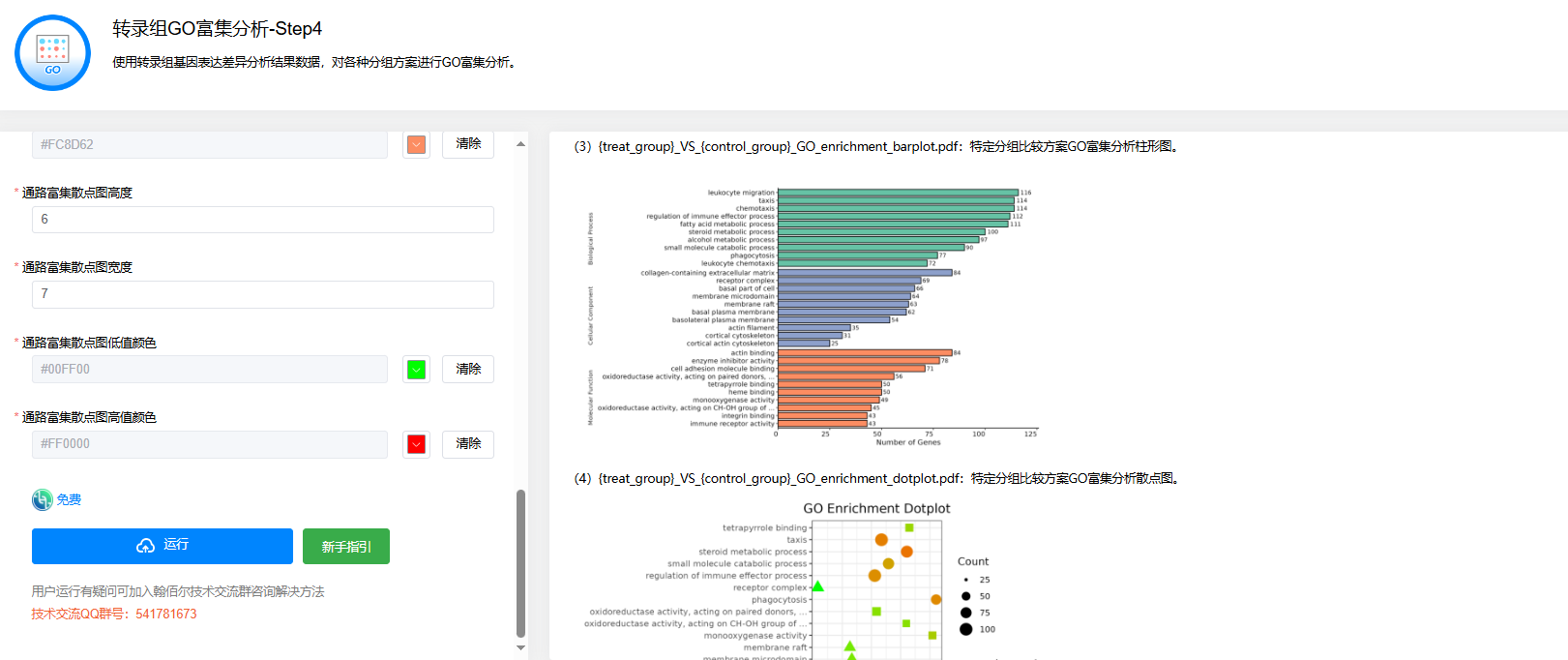
点击【转录组GO富集分析】→选择第二步【转录组基因表达差异分析】后得到的04.DiffGenesAnalysis.tar.gz文件→设置【样本分组比较方案】【物种名称】【显著性统计方法】【差异显著性统计方法】【显著性阈值】等参数→点击【运行】
输出结果为GO富集分析结果表、柱形图、散点图、气泡图
第五步:转录组GSEA分析
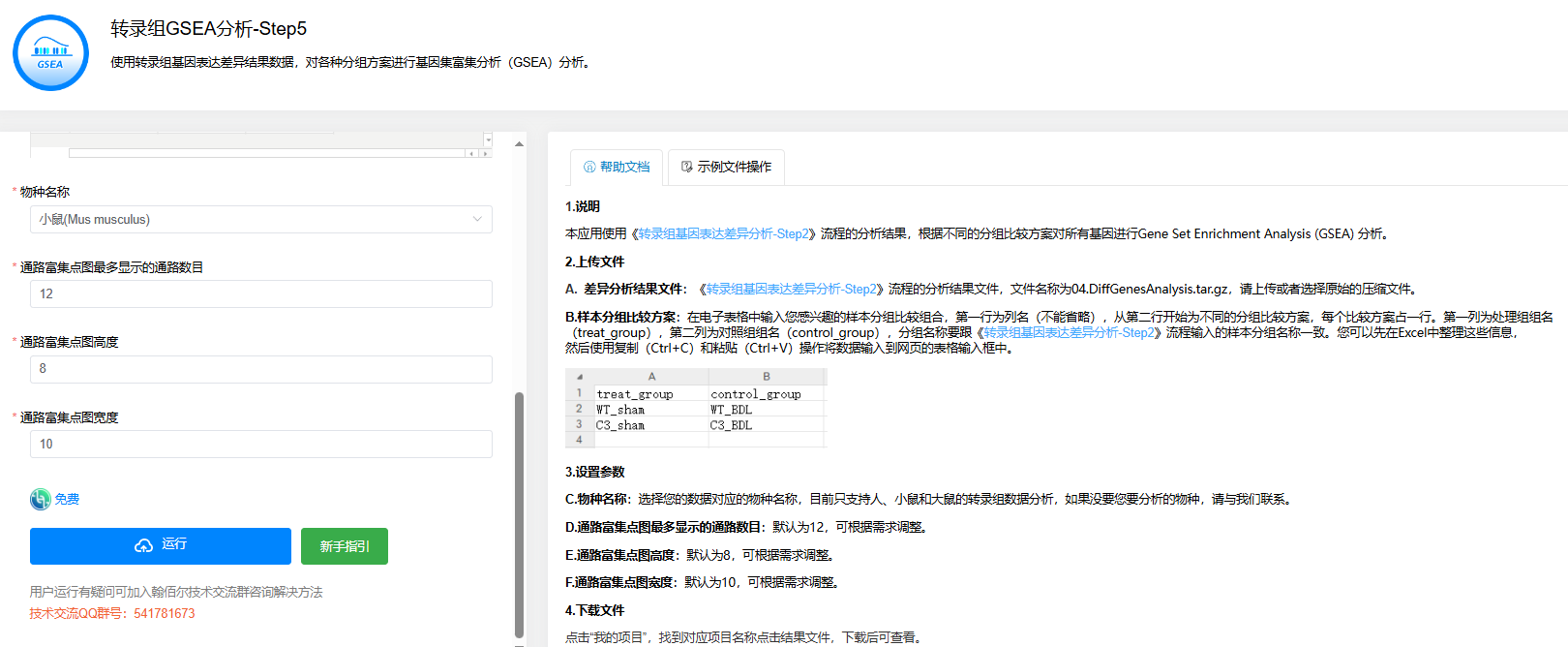
点击【转录组GSEA富集分析】→选择第二步【转录组基因表达差异分析】后得到的04.DiffGenesAnalysis.tar.gz文件→设置【样本分组比较方案】【物种名称】等参数→点击【运行】
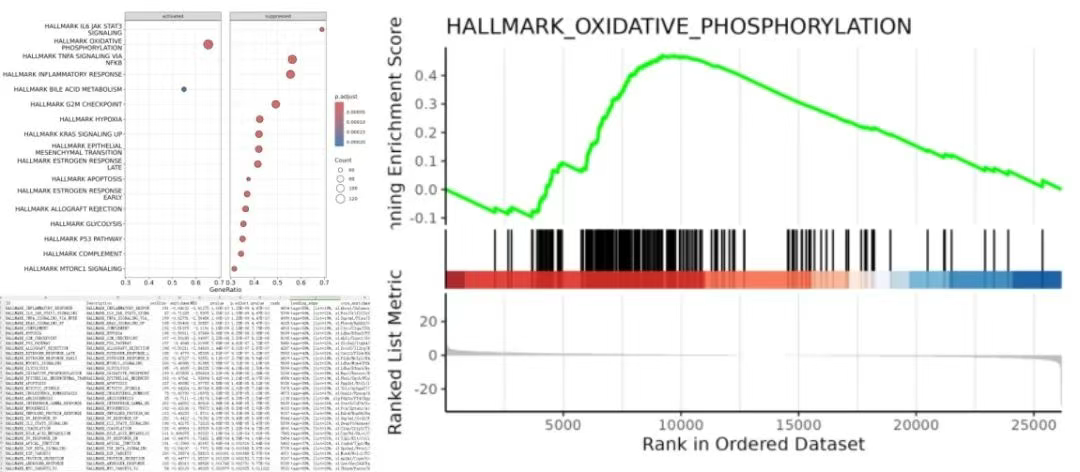
输出结果为GSEA富集分析结果表、点图、分析图
工具链接:https://www.henbio.com/tools
如果您觉得这个网站对您有帮助,还请您帮忙多多转发,与他人分享,让更多人受益。如果内容有任何侵权或是错误,恳请您及时联系我,我一定第一时间改正,感谢!

















 3677
3677

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









