







作者:哇塞
编辑:李宝珠
转载请联系本公众号获得授权,并标明来源
乔治奥古斯特大学的团队通过开发创新性的计算生物学算法 SimplifiedBondfinder,系统分析超 86,000 个高分辨率 X 射线蛋白质结构,新发现了此前从未观察到的精氨酸(Arg)-半胱氨酸和甘氨酸(Gly)-半胱氨酸之间形成的新型 NOS 键。
在细胞这个「工厂」中,氮-氧-硫(NOS)键就像一个可逆的「智能开关」,能够根据环境中的氧化还原变化调节酶活性。2021 年,来自德国哥廷根乔治奥古斯特大学的团队,通过研究淋病奈瑟氏球菌的转醛醇酶,发现了存在于赖氨酸和半胱氨酸之间的 NOS 键。这项研究超越了单一病原体和酶的研究范畴,为跨学科的蛋白质科学、药物设计和生物工程奠定了重要基础。
然而,随着蛋白质结构数据的爆炸式增长,以及科学界对蛋白质结构中化学键的持续研究,新的问题也随之而来,是否还有被忽视的其他 NOS 键或化学相互作用呢?
基于以上思考,乔治奥古斯特大学的 Sophia Bazzi、Sharareh Sayyad 团队通过开发创新性的计算生物学算法 SimplifiedBondfinder,开启了蛋白质共价键探索的新篇章。该团队通过整合机器学习、量子力学计算,构建高分辨率 X 射线晶体学数据库,系统分析超 86,000 个高分辨率 X 射线蛋白质结构,不仅新发现了 69 个 NOS 键,其中还包括了此前从未观察到的精氨酸(Arg)-半胱氨酸和甘氨酸(Gly)-半胱氨酸之间形成的新型 NOS 键。
这一革命性的发现拓宽了蛋白质化学的范围,并使药物设计和蛋白质工程中的靶向调节成为可能。与此同时,虽然该项研究的重点是 NOS 键,但这一方法同样可以灵活地运用到研究其他广泛的化学键和共价修饰中,包括结构可分解的翻译后修饰(posttranslational modifications, PTMs)。
研究成果以「Revealing arginine-cysteine and glycine-cysteine NOS linkages by a systematic re-evaluation of protein structures」为题,发表于 Communications Chemistry。
研究亮点:
* 打破了科学界认为 NOS 键仅存在于赖氨酸(Lys)-半胱氨酸之间的普遍认知,以创新性的方法首次揭示了精氨酸-半胱氨酸和甘氨酸-半胱氨酸 NOS 键的全新氧化还原调控机制
* 所提方法集成机器学习、量子力学计算和高分辨率 X 射线晶体学数据,解决了该领域研究中缺乏系统性化学键发现算法的挑战,摆脱了传统实验限制,为后续研究提供了可靠易用的工具
* 通过机器学习和人工智能技术,显著降低了此类研究的成本,同时提升了研究效率,为机器学习驱动的技术在破译蛋白质功能和识别新的蛋白质相互作用方面树立了榜样

论文地址:
https://www.nature.com/articles/s42004-025-01535-w
关注公众号,后台回复「NOS 调控」获取完整 PDF
更多 AI 前沿论文:
https://go.hyper.ai/UuE1o
数据集:多层限制提取可靠数据集
SimplifiedBondfinder 所收集的数据来自于 3 个不同的蛋白质数据库,分别是 PDB、PDB-REDO 和 BDB,收集到的数据会经过各种约束以过滤出可靠可用的数据集。其中,数据库 PDB-REDO(截止 2024 年 1 月)通过对 PDB 中的静态结构进行重新精修和优化,使其更符合当代晶体学标准,与原始的 PDB 条目相比,具有更高的准确性和可靠性。如下图左侧所示:
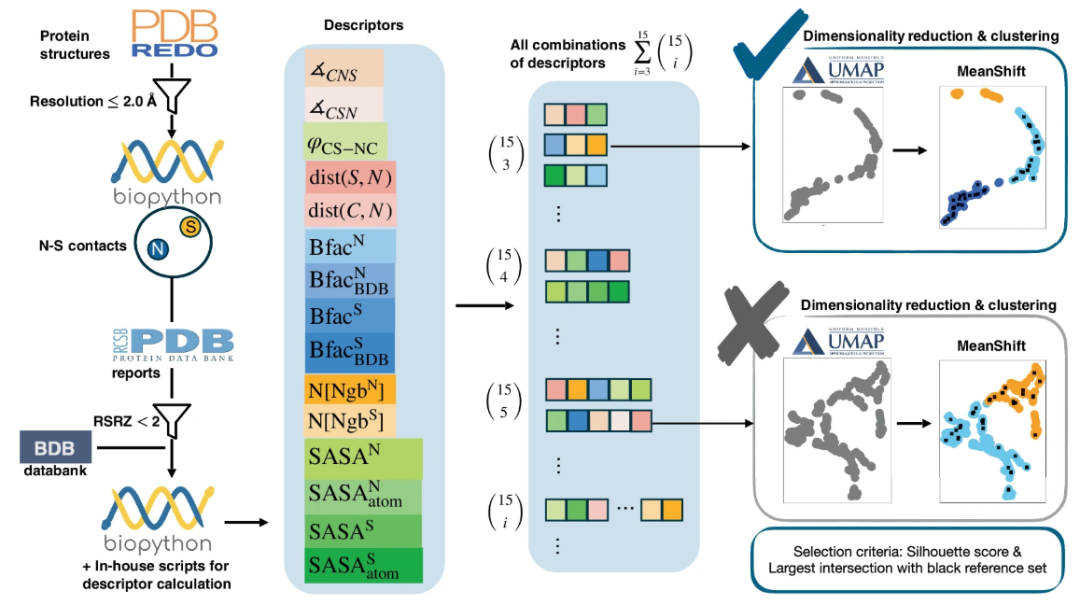
SimplifiedBondfinder 中的数据采集流程及 ML 方法
具体来看,研究团队在最初含有 170,251 个蛋白质数据的数据库中,使用多个相互关联的函数驱动自动数据集生成。其首先利用 Biopython(v 1.79)进行结构解析(使用 MMCIFParser 和 PDBParse),并计算其他原子和残基属性。经过仅解析通过 X 射线确定的结构,研究团队优化出 170,127 个蛋白质数据。
随后,为了进一步提高预测准确性,研究团队进一步筛选出分辨率 ≤ 2 Å 的蛋白质结构,最终得到 86,491 个结构用于实验分析。
为了构建用于研究特定化学键的数据集,研究团队根据组成原子类型、残基名称、原子间距离和占有率建立了标准。对于标准残基中涉及硫(S)和氮(N)原子的 NOS 连接,研究团队将 S-N 的原子间距离,即 dist(S,N),限制在 ≤ 3.2 Å,对应于赖氨酸和半胱氨酸之间供价相互作用的截止值,同时为了排除位置不确定性高的原子,将占用数阈值设置为 > 0.8。经过这一标准,研究确定了 25,462 个 N-S 接触。
为了确保所描绘的目标原子质量,研究团队进一步应用了 real-space-R-value Z-score(RSRZ),阈值设置为 <2.0,确保能够识别在真实空间中与数据可靠匹配的情况。至此,数据集进一步减少到 23,129 个 N-S 接触。这使得实验目标主要集中在半胱氨酸的两种相互作用类型上:即半胱氨酸的硫原子与甘氨酸的主链氮之间的相互作用;半胱氨酸的硫原子与精氨酸和赖氨酸的侧链氮之间的相互作用。
接下来,研究团队利用 Biopython 中的 NeighborSearch 模块提取结构参数,每个数据集中的每个样本收集 15 个不同的描述符,包括角度(∡CSN, ∡CNS)、扭转角(φCS-NC)、其他距离(dist(C, N), dist(S, N))以及利用 Bio.PDB.SASA 进一步计算得到的目标原子的溶剂可及表面积(Solvent Accessible Surface Area, SASA)值和相应的残基等。
研究团队在实验中纳入原子的 B-factors(Bfac),是为了在分析中有一个目标原子迁移率的参数,这些值来自于 2 个数据库,分别是 RCSB PDB 和一个具有一致 B-factor 的 PDB 文件数据库(BDB)。
值得一提的是,基于本研究特定要求,实验仅选择了 15 个描述符,但研究团队表示,所提算法对它可以处理的描述符数量并没有严格的限制,通过设计它可以容纳任意数量的描述符,这使它能够整合特定领域的知识或适应新的实验方法。
模型架构:融合机器学习与量子力学计算
上述部分为研究所提方法关键步骤中的第一步部分,即构建针对特定化学键的目标数据集,并应用严格的标准。本部分着重介绍所提方法的第二个关键步骤,即使用机器学习技术来探索这些高维数据,识别有效的结构描述符并预测共价键形成的潜在位点。
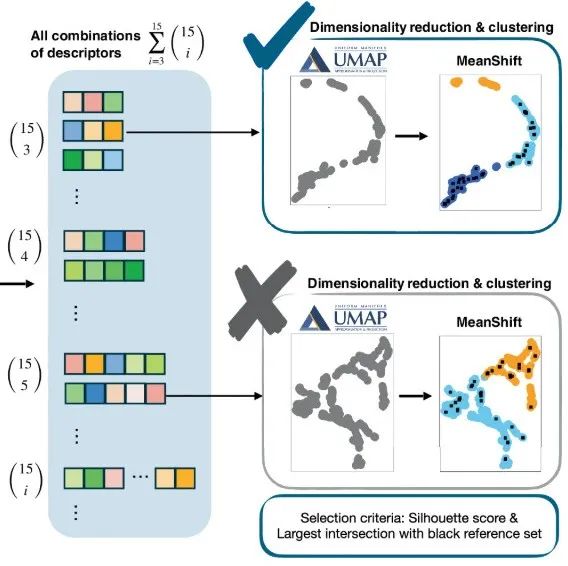
SimplifiedBondfinder 中的 ML 方法
如上图所示。首先,研究团队应用了最大嵌入维度为 3 的无监督统一流行近似与投影(Uniform Manifold Approximation and Projection, UMAP)降维技术,然后对所有可能的描述符集合进行均值漂移聚类(mean-shift clustering)。
其中,UMAP 以最佳方式保留了高维数据流行的内在拓扑和几何特性,可以确保在低维嵌入中保留基本的结构特征,便于进行有意义的下游分析。至于 UMAP 中嵌入维数的选择,则取决于数据集及其原始高维流行的拓扑和几何特性。在实际应用中,二维或三维嵌入的可解释性最强,因为它们能够实现直观的可视化,并对聚类质量进行评估。
在本研究中,3 个嵌入维度提供了良好分离且有意义的聚类,证明了选择的合理性。化学键分析和聚类结果表明,这种降维方法对于此实验的数据集来说最优,选择高于必要嵌入维度虽然能保留原始流行特征,但却会徒增计算成本而不能提升可解释性。相反,将维度降低到最优水平以下,则会导致大量信息丢失和聚类分离效果不佳。
随后,研究团队获得所有三维嵌入坐标的轮廓系数(Silhouette Score),以评估每种组合聚类质量。该算法输出聚类、轮廓系数以及每个聚类中的参考目标连接。每个候选对象通过目标原子的名称、相应的残基名称、残基编号、链和 PDB ID 来识别,以区分蛋白质内的所有目标原子。
为了找到最终且最小的特征空间,研究团队采用了多个标准,包括轮廓系数的值、每个特征空间产生的聚类数量以及这些聚类中参考目标连接的分布。
具体来说,研究团队旨在确定一个特征空间,该空间能有效地将数据分割为两个或三个不同的聚类,且轮廓系数 ≥ 0.5。在理想情况下,其中一个聚类中不包含任何参考目标连接,称为「不可能聚类」,在实践中,该聚类中参考样本的数量最少是可以接受的。其余包含所有或大部分参考目标连接的聚类称为「可能聚类」。
通过引入包含目标化学键可能和不可能候选簇,研究团队能够识别优化的特征空间,以区分可能形成新化学键的目标原子对和不太可能形成此类键的目标原子对。一旦确定一组能够可靠区分这些情况的描述符,就无需再纳入其他描述符。该方法在计算年效率和可解释性方面均有优势,可以大幅提升识别蛋白质结构内新化学键形成方面方法的预测准确性。
除了机器学习外,本次研究所提方法中还整合了量子力学计算。研究人员针对 Lys-NOS-Cys、Gly-NOS-Cys、ARG-NηOS-Cys 和 ARG-NεOS-Cys 复合物中 NOS 连接的潜在候选物进行了几何优化。使用软件包 Gaussian16 - A.03(Gaussian 16,修订版 C.01),在水中采用 B3LYP-D3 (BJ)/def2-TZVPD 理论水平进行几何优化。针对优化后的结构,实验计算了数个几何参数,包括硫原子与氮原子之间的距离(dist (S, N)),以及角度(∡CSN、∡CNS、∡NOS)。
为了验证所提聚类方法预测的 NOS 共价键的存在,研究团队使用 phenix.refine (version 1.20.1-4487-000) 对 4 个具有代表性的蛋白质结构进行了重新优化;使用 phenix.molprobity 进行了全面的结构验证,以评估几何质量、冲突分数和空间相互作用,确保与高分辨率晶体学数据一致;使用 phenix.table1 生成了完整的验证报告,总结了精修统计数据、模型质量指标和立体化学偏差。通过这些验证步骤,证实了 NOS 连接的结构完整性及其与电子密度图的兼容性。
实验结果:Arg-NOS-Cys 和 Gly-NOS-Cys 键为合理共价键
为了证明所提方法的有效性,研究团队进行了多项实验,分别探讨了机器学习技术用于描述符选择、多为描述符空间的生化意义、聚类分析以及结构和热力学验证。
使用机器学习选择描述符
研究团队首先将其应用于可能存在 Lys-NOS-Cys 连接的数据,该数据集包含 527 个赖氨酸-半胱氨酸对,还包括经过实验验证的 NOS 键。经试验确定,关键描述符为由氮原子的 B-factor(Bfac(BDB)(N)),以及赖氨酸 (Ngbᴺ)和半胱氨酸(Ngbˢ)的 Cα 原子在 4 Å 半径内的相邻残基数。
研究团队进一步将分析扩展到一个包含 313 个甘氨酸-半胱氨酸对的数据集,进而探索潜在的 Gly-NOS-Cys 连接。如下图所示。
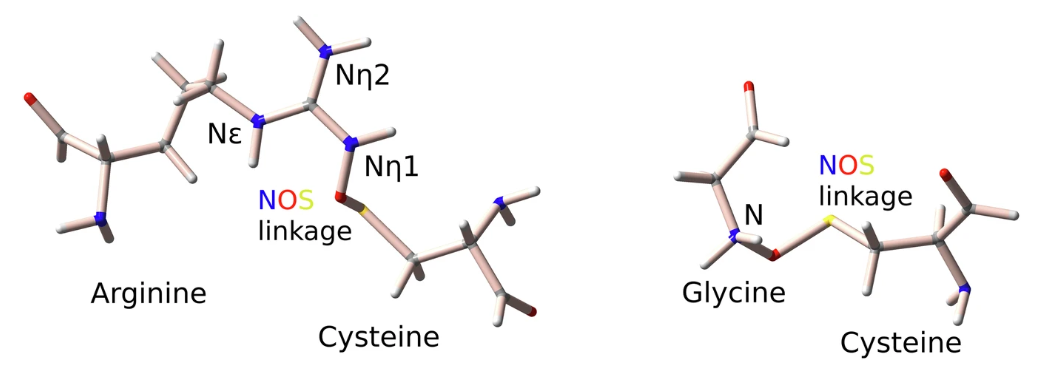
精氨酸-半胱氨酸和甘氨酸-半胱氨酸之间的 NOS 键的示意图
在此,关键描述符集包括含硫残基的 B-factor(BfacBDBS)、硫-氮距离(dist(S,N))和碳-硫-氮角度(∡CSN)。
在预测精氨酸和半胱氨酸残基之间形成 NOS 键的关键描述符方面,精氨酸侧链有Nη 和 Nε 两种类型的氮原子,它们在几何特征和化学性质上有所不同,因此研究分别分析了 Nη (Arg-NηOS-Cys) 和 Nε (Arg-Nε-Cys) 的数据集。
对于 Arg-NηOS-Cys,所选的描述符符合包含氮残基(SASAᴺ)的溶剂可及表面积、∡CSN 以及与硫相邻的残基(Ngbˢ)和与氮相邻的残基(Ngbᴺ);同样对于 240 个 Arg-NεOS-Cys 对的数据集来说,关键描述符涉及 BfacBDBS、SASAˢ、氮原子的溶剂可及表面积、∡CSN 和 ∡ CNS。
这些发现通过 UMAP 降维可视化显示出清晰的聚类分离,如下图所示,其中天蓝色和宝蓝色表示为 NOS 键候选,橙色则表示为「不可能聚类」,黑色方点为参考数据集,由此清晰可见,可能形成 NOS 键的样本与参考标准点的分布高度重合。
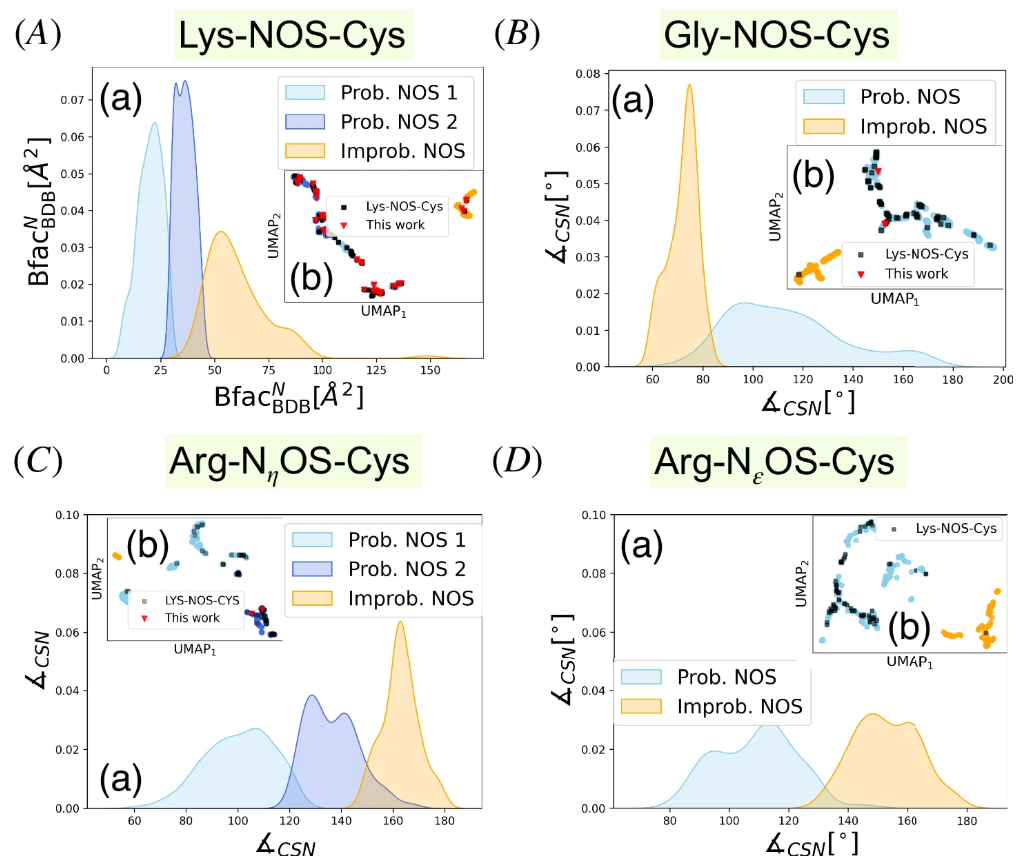
SimplifiedBondfinder 得到的密度分布图和 UMAP 结果
多维描述符空间的生化意义
研究团队探讨了关键描述符的生化相关性。通过算法确定最小描述符集,关键描述符对区分 NOS 和非 NOS 键具有重要意义。
以 B-factor 为例,不同聚类中 B-factor 呈现出不同的分布模式,如上 A(a)中所示的那样,对于「可能聚类」和「不可能聚类」,B-factor 的众数并不相同,且 B-factor 与原子或区域的灵活性相关,活性位点残基通常 B-factor 较低,说明了其与酶活性有关。不过研究团队同样指出,低 B-factor 可能指示 NOS 键合,但也可能反应其他的氮-硫相互作用。
针对不同氨基酸残基形成的 NOS 键描述符特点,BfacBDBᴺ 在 Lys-NOS-Cys 中是区分两类聚类的主要因素;针对 Gly - NOS - Cys 连接,∠CSN 是区分可能的 NOS 连接聚类的主要描述符,多数可能样本的 ∠CSN >80°,优化的 Gly - NOS - Cys 复合物的∠CSN值约为 94°;∠CSN 仍是针对 Arg - NεOS - Cys 连接区分可能与不可能的 NOS 连接的关键决定因素。
聚类分析
在本环节评估中,研究团队检测到了 65 个 Lys-NOS-Cys 键、2 个 Gly-NOS-Cys 键(下图 a 和 b)和 2 个 Arg-NηOS-Cys 键(下图 c 和 d)。
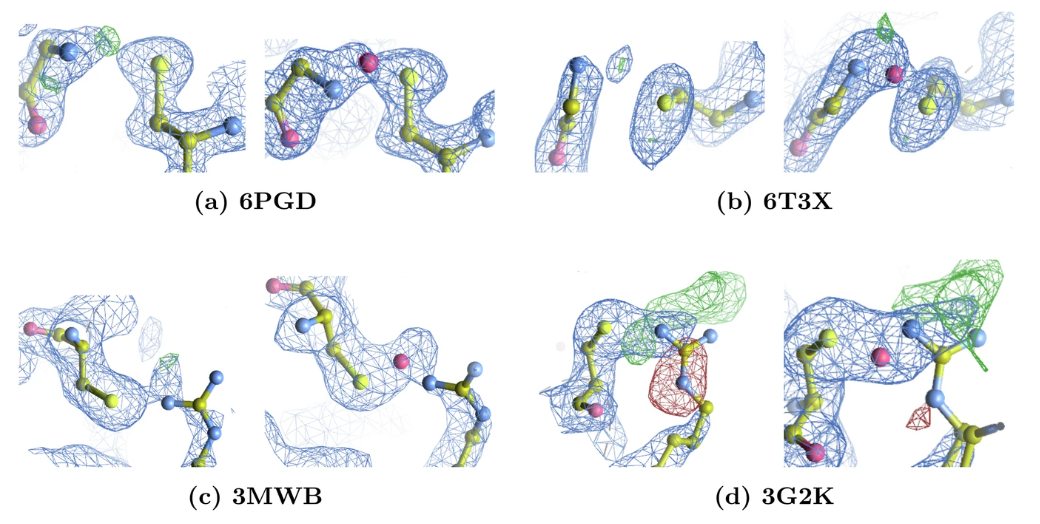
预测 NOS 键细化前后的电子密度对比
研究团队通过明确建模和重新精修,引入 NOS 键后 Rwork / Rfree 值平均改善 0.5%,未解释的电子密度峰显著减少。对于 3G2K,原始结构中精氨酸侧链周围有负电子密度峰,重新分配精氨酸构象后显著减小,且两个模型中精氨酸侧链附近都有正差异峰,因其幅度大且存在 DMSO,可能代表当前模型中未建模的溶剂分子。
结构和热力学验证
研究团队为进一步证实 Arg-NOS-Cys 和 Gly-NOS-Cys 之间的联系,将量子力学几何优化与 4 个代表性蛋白质复合物(6PGD, 6T3X, 3MWB, and 3G2K)的热力学评估结合起来,从而系统地解释体内可能存在的化学变异性。
在结构验证方面,在 NOS 键优化模型中,S-N 距离范围为 2.61 ~ 2.70 Å,与原始 PDB-REDO 结构的 2.63 ~ 2.89 Å 区间非常接近。而去除桥接氧原子的模拟导致 S-N 分离显著增加,达到 3.36-4.26 Å,这表明了实验观察到的 S-N 距离较短与中间氧原子的存在一致。
在热力学评估方面,研究团队计算了不同质子化状态下的吉布斯自由能(ΔG),显示所有 NOS 键形成过程中都是负值。这表明在模拟状态下,用氧取代一个氢形成 NOS 键在热力学上是可行的。然而,ΔG 的大小随质子化状态以及精氨酸和甘氨酸衍生的复合物之间存在显著差异。在这两个体系中,中性的甘氨酸或精氨酸比带正电荷的状态更受青睐。基于甘氨酸的复合物表现出略高的 ΔG 值。虽然这些值仍意味着在热力学上有利的联系,但它们在系统上比相应的精氨酸复合物放能少。
总而言之,这些结构结果提供了一致的证据,表明了 Arg-NOS-Cys 和 Gly-NOS-Cys 键是合理的共价键,而不是简单的非键接触。同时,量子力学优化的几何形状和晶系的晶体学数据之间的一致性,以及负自由能的形成,有力地表明这些连接在相关蛋白质环境中,无论是在结构上还是能量上都是可行的。
机器学习打开蛋白质微观世界新篇章
正如论文中所提到,快速发展的机器学习和人工智能技术在解决生物化学中的复杂问题方面,已经展现出了超越传统生物化学方法的优越性,它以低廉的计算成本和高效的方式,促使着科研界展开了一场关于「生产方式」的大变革,也推动者机器学习驱动的技术在破译蛋白质功能和识别新的蛋白质相互作用发挥更大的潜力。
无独有偶,如美国加州理工学院的 Kevin K.Yang 等人发表于 Nat. Methods,题为「Machine learning-guided directed evolution for protein engineering」的文章,通过对比定向进化和机器学习辅助定向进化,阐述了机器学习的优越性。同时文中还列举了如酶催化效率、细胞色素 P450 热稳定性优化等实际案例,提到线性回归、高斯过程、贝叶斯优化等多种机器学习方法,表明了机器学习可为蛋白质工程提供「数据驱动的智能导航」,通过建模序列-功能关系,显著提升定向进化的效率和成功率。
论文地址:
https://arxiv.org/pdf/1811.10775
另外,意大利博洛尼亚大学的 Rita Casadio 等人以「Machine learning solutions for predicting protein–protein interactions」为题发表的文章,同样详细介绍了机器学习在蛋白质研究方面的探索。其中介绍了包括无监督和有监督学习的机器学习方法在蛋白质-蛋白质分子相互作用(PPI)中的应用,重点突出了其在数据质量、表示、训练算法和验证程序等方面的关键问题。
论文地址:
https://wires.onlinelibrary.wiley.com/doi/full/10.1002/wcms.1618
总的来说,在蛋白质的微观世界中,仍有诸多关乎生命的密码隐藏其中,而机器学习为主要手段的系统性数据驱动的方法无疑就像一把打开蛋白质微观世界大门的钥匙,激发着科研界对蛋白质功能、稳定性进行更深入的研究和探索,从而不断破除人类对生命的认知局限。

往期推荐





戳“阅读原文”,免费获取海量数据集资源!


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









