






Introduction
除了miRNA表达数据,各种miRNA相关的知识也强有力地支持了对miRNA功能相互作用的理解。
那些具有许多共同调控靶基因或疾病的miRNAs可能具有相似的功能
一些方法通过考虑实验验证的miRNA-靶标关系来评估miRNA相互作用,评估miRNA功能相互作用的直观方法是通过Jaccard指数(Jaccard index)计算共同靶标的比例。但是,它丢失了大量的相似信息
杰卡德系数(Jaccard Index)
杰卡德系数,又称为杰卡德相似系数,用于比较两个样本之间的差异性和相似性。杰卡德系数越高,则两个样本相似度越高。
一些方法尝试使用基因本体注释或者)蛋白质-蛋白质相互作用(PPI)作为推断miRNA相互作用的靶基因的额外信息。
然而,GO数据库对大量靶基因的注释信息有限,不利于准确评价miRNA相互作用。同时,PPI数据库中存在大量不符合实验有效性的假阳性,这可能会给计算miRNA功能相似性带来偏差。
"Gene Ontology Annotations"(基因本体注释)是一种用于描述基因和蛋白质功能的标准化方法。基因本体是一个系统化的生物学术语表,它将基因和蛋白质的功能划分为三个主要的本体(ontology):分子功能(molecular function)、细胞组分(cellular component)和生物过程(biological process)。
总的来说,Gene Ontology Annotations 提供了一种标准化的方式来描述基因和蛋白质的功能,使得科研人员能够更好地理解生物体内各种生物学过程。
"Protein–protein interactions"(PPIs,蛋白质-蛋白质相互作用)是指在生物体内,两个或多个蛋白质之间发生的相互作用或结合。这种相互作用对于维持细胞结构、调控信号传导、执行代谢途径等生物学过程至关重要。
总结:
miRNA与疾病的关系提供了miRNA参与疾病发生发展的直接证据,成为评价miRNA功能相互作用的有力选择。
一种miRNA通常调控多种疾病的发生发展过程,一种疾病与多种miRNA相关。基于疾病的miRNA功能相似度计算的关键是准确评估疾病语义相似度。
Wang等人[23]提出了一种基于疾病有向无环图(DAG)的疾病语义相似度图推理方法。然而,基于DAG的疾病相似性通过平等地评估与根病具有相同距离的疾病的重要性而忽略了特定疾病的语义意义[24]。
因此,要获得高质量的miRNA功能相互作用,必须合理评估和充分利用miRNA-疾病关系中的疾病语义信息。
同时,利用miRNA表达数据和知识库中的信息,有利于构建稳健的miRNA相互作用网络。miRNA表达数据包含了不同样本组之间miRNA协同调控的变化信息。已知的miRNA与疾病的关系为miRNA功能相似性的评价提供了有力的支持。因此,将miRNA协同调控与miRNA功能相似性相结合,可以促进下游网络分析任务的开展,包括潜在的疾病生物标志物的鉴定。
定义基于分子相互作用网络的信息子网络是生物网络分析中的一个重要课题。一些方法采用聚类技术来划分生物网络。一般来说,需要预定义的簇号,但很难确定。一些方法采用启发式策略来识别重要模块。
Zhang等人[26]提出了基于网络的博弈论方法(NGTM),通过在基于博弈论的模块扩展中使用合作博弈论度量(Shapley值)评估特征贡献来识别潜在的癌症子网络生物标志物。但基于启发式的模块识别方法所使用的启发式信息有限,容易陷入局部最优。
与监督学习和无监督学习不同,强化学习(RL)旨在做出最大化长期回报的决策[27]。因此,RL策略可以通过充分探索解空间以获得全局最优结果来为模块生物标志物识别带来更多可能性。
Paim等人[28]尝试使用RL来检测复杂网络中的社区,并提出了Q-Learning [29] for Community Detection(QLCD)方法。网络中的每个节点充当一个代理,从其最近的邻居节点(动作空间)中选择一个节点组成集群。代理节点学习的行动策略,以最大限度地提高网络的模块化。然而,由于QLCD固有的简单动作空间和学习策略不足,在疾病研究中可能无法找到竞争模块。有必要进一步探索强化学习在定义疾病网络生物标志物方面的潜力。
为了有效识别潜在的miRNA疾病生物标志物,提出了基于多视图网络和强化学习的miRNA数据分析方法miRMarker。
- 基于表达数据构建miRNA协同调控网络。
- 利用公共知识库中已知的miRNA-疾病关系构建miRNA功能相似性网络。
- 然后,miRMarker整合两个miRNA网络,并通过强化学习策略定义关键的miRNA模块。
我们通过在9个转录组学数据集上与8种有效的数据分析方法进行比较,验证了miRMarker在疾病样本区分方面的有效性。
此外,我们检查了由miRMarker定义的结直肠癌的潜在miRNA模块生物标志物。实验结果表明miRMarker在确定疾病诊断和预后的重要模块生物标志物方面具有巨大的潜力。
材料和方法
miRMarker由两个主要部分组成:
(i)分别基于miRNA表达数据和miRNA-疾病关系构建miRNA网络,整合两个网络;
(ii)通过强化学习策略定义关键的miRNA模块。图1显示了miRMarker的工作流程。
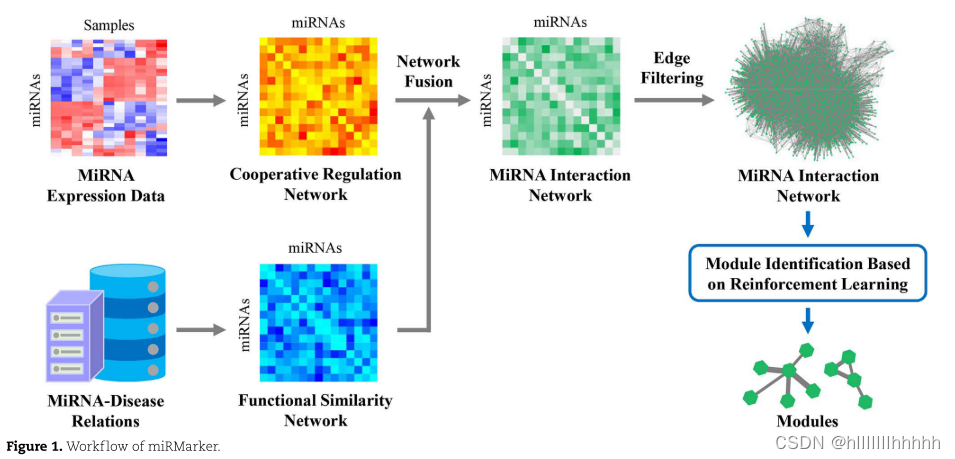
miRNA表达数据集
在这项研究中,收集了9个miRNA表达数据集,以评估miRMarker的有效性。所有数据集均来自公共数据库Gene Expression Omnibus(GEO),涉及多种疾病,如肝细胞癌、血小板增多症和结直肠癌。
表1给出了9个miRNA表达数据集的详细信息。数据集GSE 41574、GSE 67139、GSE 32273、GSE 34496、GSE 41282和GSE 108153是两类数据集。数据集GSE 31164、GSE 39046和GSE 35834是多类数据集。
我们使用miRBase v22.0将所有人类成熟miRNA名称映射到标准miRNA编号。将代表相同miRNA的探针的表达值平均。
因为同一个miRNA可能会被多个探针检测到,而这些探针的测量结果可能会有一些变化。通过取平均值,可以得到更为稳定和可靠的miRNA表达量。
miRNA与疾病的关系
从两个最大的知识库miRCancer [20]和miR 2Disease [21]中提取手动策划的miRNA-疾病关系。
miRCancer使用文本挖掘技术从PubMed数据库中的医学文献中提取miRNA-癌症关联,然后手动修改关联。
"miR2Disease提供了从已发表的论文中整理出的miRNA与人类疾病之间的全面调控关联信息。
我们下载了miRCancer(9080个条目,于2022年6月下载)和miR2Disease(2877个条目,于2022年7月下载)的最新版本。
所有人类成熟miRNA的名称都通过miRBase映射到标准miRNA存取号。将关系中的疾病名称映射到MeSH中的规范疾病术语(于2022年7月下载)。
我们整合了miRCancer和miR 2Disease的miRNA-疾病相互作用,并消除了重复条目。最终获得了6099个miRNA与疾病的关系,涉及163种人类疾病。
miRNA网络构建
miRMarker通过整合协同调控网络GCR和功能相似性网络GFS构建了miRNA相互作用网络。使用miRNA表达数据构建GCR。GFS是基于miRNA与疾病的关系建立的。
设F = f1,f2,...,fm表示特征(miRNA)集,m是特征的数量。miRMarker定义了两个miRNAs fi和fj(1≤ i < j ≤m)的组合特征fij = fi − fj,代表fi和fj之间的相对表达关系。
fij从正常样本到疾病样本的表达水平失调描述了fi和fj协同调节的变化。采用Mann-Whitney U检验测量两个样本组之间fij的差异显著性(多类样本的Kruskal-Wallis检验)。
然后,fi和fj之间的边的权重被定义为U检验中fij的P值(P-valueij)的负对数值。如果两个不同样本组之间fij的差异显著,则fi和fj之间的边权重较大。这意味着在GCR中fi和fj之间的协同调节关系是强的。
定义1.协同调节网络
设GCR = {VCR,ECR,WCR}是一个协同调节网络.
VCR = F是顶点集,
ECR = {(fi,fj )| fi,fj ∈ F,1 ≤ i < j ≤ m}是边集,
WCR ={ w( fi,fj )=−log(p-valueij)|( fi,fj )∈ ECR}表示边权重集。
miRMarker构建了基于miRNA-疾病关系的功能相似性网络GFS。
图2显示了fi和fj之间miRNA功能相似性的计算过程。
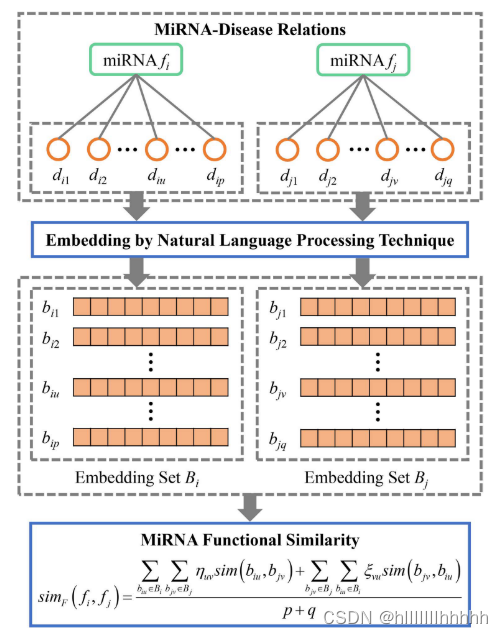
miRNA对应于包含许多人类疾病的受调控疾病集。
fi和fj之间的miRNA功能相似性定义为相应的受调控疾病集
Di ={di1,di2,.,dip}
Dj={dj1,dj2,...,djq}
diu(1≤u≤p)是第u个受fi调节的疾病。
djv(1≤v≤q)是fj调节的第v类疾病。
p和q分别是Di和Dj中的疾病数量。
我们采用自然语言处理(NLP)模型BioBERT [32]将疾病术语嵌入数字向量。BioBERT是一个强大的NLP模型,在大量的生物和医学语料库上训练。该算法能够生成高质量的生物医学文本嵌入向量。疾病嵌入向量编码了疾病的判别语义信息
通过在Di和Dj中嵌入疾病,我们得到了嵌入集
Bi ={ bi 1,bi 2,.,bip }
Bj= {bj1,bj2,.,bjq}
biu(1≤u≤p)是diu的嵌入向量。
bju(1≤v≤q)是djv的嵌入向量。
如果diu和djv相似,则两个对应的嵌入向量biu和bjv之间的相似性高。所有疾病对(Di和Dj之间)的余弦相似度值构成了一个p × q的疾病语义相似性矩阵MD。MD的第u行和第v列中的元素指示biu和bu之间的余弦相似度值。采用加权最佳匹配平均策略,通过对MD中每一行和每一列的相似性值进行加权平均,来聚合疾病相似性值。fi和fj之间的miRNA功能相似性定义如下:
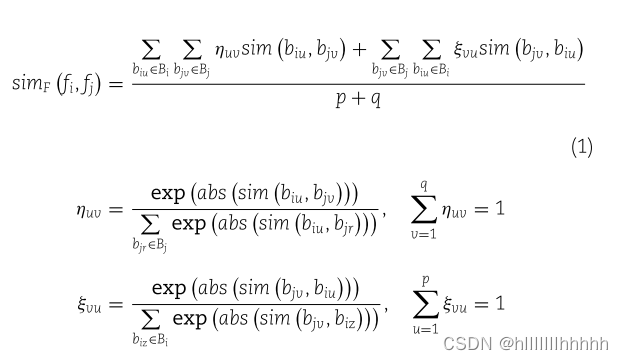
其中,sim(biu,bjv)是biu和bjv之间的余弦相似度值。ηuv和εvu分别是行归一化和列归一化的加权因子。大的sim(biu,bjv)导致大的加权因子ηuv。abs()表示绝对值函数。miRNA功能相似性的计算simF(fi,fj)考虑了Di和Dj中每对疾病之间的相似性。如果疾病集合Di和Dj之间的相似性高,则fi和fj之间的功能相似性高。
定义2.功能相似性网络
设GFS = {VFS,EFS,WFS}是一个功能相似网络.
VFS = F是顶点集,
EFS = {(fi,fj)| fi,fj ∈ F,1 ≤ i < j ≤ m}是GFS的边集,
WFS ={ w(fi,fj)= simF( fi,fj )|(fi,fj )∈ EFS}表示边权重集。
miRMarker通过整合协同调控网络GCR和功能相似性网络GFS定义了miRNA相互作用网络GFN。GCR和GFS都是无向完全图。GCR和GFS中的所有边权重分别使用最小-最大归一化进行归一化。
在GFN中,(fi,fj)的边权重是GCR和GFS中归一化边权重的平均值:
![]()
其中,w'CR(fi,fj)和w'FS( fi,fj)分别是GCR和GFS中的(fi,fj)的归一化边权重。GFN是一个包含许多低权无信息边的完全图。miRMarker通过保留信息边来定义GFN的子网络。首先执行最大生成树(MST)算法[33]以确保网络连通性。
GFN的最大生成树GFN−MST仅包含最大化边权重总和的m−1条边。miRMarker将GFN−MST中的每个节点链接到其k个最近邻(最大边权重)以获得最终miRNA相互作用网络GFN−MST−kNN。它是一个连通的、富含miRNA相互作用信息的网络,具有高权重的边。
miRNA模块鉴定
miRMarker利用基于值的强化学习(RL)方法Q-Learning [29] 从GFN−MST−kNN中识别潜在的miRNA模块生物标志物。它通过维护代理的Q表找到最优的动作选择策略。Q表记录了在每个状态下采取不同动作所获得的最大长期奖励(Q值)。Q-Learning在探索和与环境交互过程中更新Q值,旨在通过根据Q表采取一系列最优动作来最大化未来奖励。
miRMarker在GFN−MST−kNN中选择具有最高综合重要性得分的g个节点作为初始状态节点。fi(1≤ i ≤m)的综合重要性通过结合个体的区分能力和网络拓扑重要性来衡量,具体如下:
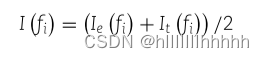
其中,
Ie(fi) = -log(p-valuei) 是fi的归一化表达差异分数,表示在疾病样本区分中的个体区分能力。
- p-valuei 是与fi相关的p值,表示在统计检验中衡量表达差异的概率。p-valuei 是fi在Mann–Whitney U检验(对于多类样本的情况为Kruskal–Wallis检验)中的P值。
- 如果fi的表达值在不同样本组之间显示显著差异,这意味着fi具有较高的个体区分能力。
It(fi)= ClosenessCentrality(fi)是fi的归一化网络拓扑重要性得分,定义为fi在GFN−MST−kNN中的中心度。
- 中心度测量一个节点到网络中所有其他节点的距离。
- 如果fi的中心度较大,意味着fi在GFN−MST−kNN的中心位置,fi的网络拓扑重要性得分较高。
Ie(fi)和It(fi)都使用最小-最大归一化进行归一化。节点的综合重要性得分考虑了个体区分能力和网络拓扑特性,综合评估了节点的重要性。最重要的g个节点被选为初始状态节点。
对于初始状态节点ft(1≤ t ≤m),miRMarker创建一个Q表Qt,其中所有的Q值都设为零,并且有一个包含ft的访问节点集合St。Qt的行和列分别表示状态节点和动作节点。
在Qt中,Qt (fs, fa )表示当状态节点为fs时,选择动作节点fa的Q值,即累积奖励。Q值越大,表示获得的奖励越多。miRMarker从动作空间中选择一个动作节点,即St中访问节点的邻居节点集。
为了处理探索–利用问题,采用了epsilon–greedy策略。当当前状态节点是fs时,以概率ε会随机从动作空间中选择一个动作节点。否则,miRMarker会选择导致Qt( fs, fa )最大的动作节点fa。ε的值在每个episode结束后逐渐减小,具体方式如下:
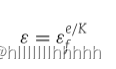
其中,εf 是一个小的值,通常设为 1e−05 [28]。e 是当前的episode数。K 表示最大的episode数。在早期的episodes中,miRMarker充分探索动作空间,倾向于选择一个随机的动作节点。ε 随着 e 的增加呈指数递减。miRMarker逐渐减少选择随机动作节点的概率,而更倾向于选择最优动作节点,即导致在动作空间中具有最大 Q 值的节点。被选择的动作节点 fa 被添加到 St 中。
将fa添加到St的奖励值同时考虑St的分类性能和大小,如下所示:
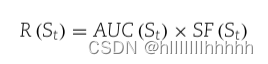
其中,
- AUC(St) 是St在ROC分析中的AUC值。
- SF(St) = 1 + sqrt(1/ |St|) 表示集合St的尺寸因子。
- sqrt() 是平方根函数。
- |St| 表示集合St的大小。
如果St具有较高的分类性能和较小的大小,奖励值 R(St) 就会很大。
Qt (fs, fa) 的更新如下:
![]()
其中,α是学习率,γ是折扣率。
如果α较大,表示与环境互动学到的信息起到主要作用,Qt ( fs, fa)会有较大的更新。
如果α较小,Qt (fs, fa)就会得到较少的更新。
max(Qt(fa, f'a))表示在状态节点为fa时选择任何动作节点f'a 的最大 Q 值。
对于 Qt( fs, fa )的更新同时利用了即时奖励、折扣最大预期累积奖励以及当前对 Q 值的估计。fs 使用最新的动作节点 fa 进行更新。然后,迭代选择新的动作节点。
当前的episode在 AUC(St) 达到最大值1.000或 |St| 超过预定义的限制时停止。在所有的episodes结束时,miRMarker已充分探索了miRNA网络并学到了用于最优动作策略的有价值信息。
miRMarker利用学到的Q表定义了关键的miRNA模块。模块扩展过程从每个初始状态节点ft开始。最初,模块Mt仅包含ft。使用基于学习的Q表的epsilon–greedy策略,迭代选择最优动作节点fa。所选的动作节点fa被添加到Mt中,状态节点也被更改为fa。
模块扩展有两种停止条件:
(i) 模块Mt的AUC达到最大值1.000,表示Mt的性能没有进一步改善
(ii) 在模块扩展期间已向Mt添加了新的动作节点fa。在这种情况下,不会添加新的节点。
算法1展示了miRMarker中模块识别的伪代码。
- 第1至2行分别初始化访问节点集合St和Q表Qt。
- 第3行初始化一个episode。
- 第4行设置当前episode的ε。
- 第5行开始搜索过程。
- 第6行定义状态节点。
- 第7行通过epsilon–greedy策略选择动作节点。
- 第8行将动作节点添加到St。
- 第9行评估St的分类性能。
- 第10行计算奖励值。
- 第11行更新Q表。
- 第12至14行判断搜索的停止条件。
- 第17行基于学到的Q表识别模块。

miRMarker为每个g个初始状态节点识别一个模块。对于每个已识别的模块,建立一个带有线性核的支持向量机(SVM)分类器[34]。采用加权投票法来整合g个分类器的预测概率值。g个分类器的权重是通过在训练样本上进行内部5折交叉验证中模块的归一化平均AUC来定义的。然后,miRMarker通过对g个分类器的加权投票为新样本进行标记。
涉及统计学
在所提出的方法中使用了多种统计学方法。
在构建合作调控网络时,采用Mann–Whitney U检验来衡量组合特征fij在两个不同样本组之间的差异显著性(对于多类别样本采用Kruskal–Wallis检验)。
在构建功能相似性网络时,采用余弦相似度来计算两个疾病嵌入向量之间的疾病语义相似性。在模块识别中,采用Mann–Whitney U检验来计算特征对于定义初始节点的个体区分能力(对于多类别样本采用Kruskal–Wallis检验)。两个组之间特征表达水平的显著差异表明该特征具有较高的个体区分能力。
实验设置
miRMarker 与7个方法比较
多变量特征选择SVM-RFE、基于网络的数据分析方法WGCNA、DNB、GroupBN 、NGTM、QLCD和定义疾病相关模块(DDRM)
- SVM-RFE集成了SVM和递归特征消除以选择信息丰富的特征
- DNB是为时间序列数据分析提出的。采用了DNB来分析静态数据。首先采用分层聚类方法获取潜在的特征组。然后,DNB分别在每个样本组上构建了Pearson相关网络,并通过评估分子相关性和不同相关网络之间的标准差来计算每个特征组的关键指数(CI)分数。具有最大CI(Causal Inference)分数的特征组被定义为DNB生物标志物。
- Group BN采用分层聚类技术将相似的特征分组,并构建贝叶斯网络来模拟特征组之间的关系以进行概率推理。
- DDRM通过同时考虑共调控的靶基因和非共调控的靶基因来评估miRNA的协同关系,然后构建加权miRNA协同网络。利用核聚类方法发现了200个miRNA功能模块。DDRM将相应的miRNA表达数据映射到miRNA功能模块,根据映射的miRNA评估每个模块,并为疾病定义关键模块。我们采用在训练样本的内部5折交叉验证中具有最大平均性能的前五个功能模块作为DDRM生物标志物。
- NGTM是为已知的基因调控网络提出的。QLCD是为现有的复杂网络数据集设计的。对于NGTM和QLCD,我们基于miRNA数据集使用对称不确定性(SU)构建了相关网络。SU是一种广泛使用的基于熵的相似性度量。每个节点与具有最高SU值的k个邻居相连。搜索k值的范围为{1, 3, 5, 7, 9},以使SU网络中的节点度最适合幂律分布。
采用了自然语言处理(NLP)技术,获取疾病语义向量,基于已知的miRNA-疾病关系计算miRNA的功能相似性。将miRMarker与使用基于疾病DAG的miRNA功能相似性网络(命名为NDAG)的方法进行了比较,旨在展示miRMarker中miRNA功能相似性计算的有效性。
- miRMarker的有效性分别在二元分类和多类分类中进行了评估,以区分不同的样本组。
- 进行了接收者操作特征曲线(ROC)分析来评估分类性能(多类分类的宏平均一对多ROC)。
- 进行了10次10折交叉验证,以获得平均曲线下面积(AUC)、灵敏度和特异性。
数据集GSE32273包含来自三种不同组织的样本,因此,它被分成三个子集,具体取决于组织类型。对于二元分类,我们还使用了多类别数据集(GSE31164、GSE39046和GSE35834),将它们分为多个二元子集。因此,在二元分类中总共包括了17个二元数据集/子集。
对于所有的特征选择方法,采用了带有线性核的支持向量机(SVM)作为分类器。在训练模型之前,对特征进行了单位方差(UV)缩放,以进行无量纲化处理。(保证数值范围一致)
所有的方法都是在R 4.2.0中实现的。
结果和讨论
miRMarker的性能评价
将miRMarker与八种高效的数据分析方法(SVM-RFE、WGCNA、DNB、GroupBN、NGTM、QLCD、N-DAG和DDRM)进行了ROC分析的比较。
- 表2显示了二元分类中AUC的比较结果。粗体字标记了相应数据集上的最高AUC值。
- 标记‘∗’表示该方法的AUC值与miRMarker的AUC值存在显著差异(t检验,P值 < 0.05)。
- ‘Average’行显示了每种方法在17个二元数据集上的平均AUC值。
- ‘Win/Tie/Loss’行列举了miRMarker的平均性能显著高于、与之不显著不同或显著低于比较方法的数据集数量。
miRMarker在大多数情况下表现优于其他比较方法。与SVM-RFE相比,在17个数据集中的AUC上,miRMarker在10个数据集上表现更好。与基于网络的数据分析方法相比,miRMarker分别在10、14、12、16、13和8个数据集上击败了WGCNA、DNB、GroupBN、NGTM、QLCD和DDRM。miRMarker在7个数据集上优于NDAG,并在10个数据集上与其并列。可以看出,miRMarker在17个数据集中的9个中获得了最高的AUC值(用粗体字标出)。
此外,miRMarker在不同数据集上获得了最高的平均AUC(0.914)和最低的平均标准差(SD)(0.110)。
在线附录表格S1和S2,可在http://bib.oxfordjournals.org/找到,分别展示了灵敏度和特异度的比较结果。可以看出,miRMarker在八个数据集上分别获得了最高的灵敏度和特异度。此外,miRMarker在数据集上获得了最高的平均灵敏度(0.948)和特异度(0.922)。
此外,我们在Matthews相关系数(MCC)上检验了miRMarker与其他数据分析方法的比较结果(表S3可在http://bib.oxfordjournals.org/找到)。miRMarker在数据集上获得了最高的平均MCC(0.662)。二元分类的比较结果说明了miRMarker的有效性。
在线附录表格S4(http://bib.oxfordjournals.org/)提供了多类别分类的比较结果。多类别分类的比较结果也表明,miRMarker比其他方法表现更有优势。
miRMarker整合了基于表达数据和已知miRNA-疾病关系构建的多视图miRNA网络,实现了强健的miRNA融合网络。通过强化学习策略定义了重要的miRNA模块。简而言之,miRMarker能够充分利用表达数据和miRNA-疾病关系中隐藏的miRNA相互作用信息,并定义了用于疾病诊断和预测的重要miRNA模块生物标志物。
消融研究
miRMarker通过整合合作调控网络和功能相似性网络构建miRNA相互作用网络。
将miRMarker与仅使用合作调控网络(命名为N-CR)或功能相似性网络(命名为N-FS)的方法进行比较,以验证网络整合的有效性。
在线附录图S1(http://bib.oxfordjournals.org/)展示了在数据集上的平均AUC值的比较结果。miRMarker的性能优于N-CD和N-FS。
合作调控网络和功能相似性网络的整合充分利用了多视图miRNA相互作用,得到了强健的miRNA相互作用。这有助于找到疾病样本判别的关键信息。
参数敏感性分析
miRMarker中有两个主要参数:
(i) 学习率 α
(ii) 初始状态节点数 g(模块数量)。
α 的设置涉及到模块识别中 Q 值的更新范围。在线附录图S2A(http://bib.oxfordjournals.org/)展示了miRMarker在不同α设置({0.2, 0.4, 0.6, 0.8})下的平均AUC值。
可以看出,当α从0.2变化到0.8时,miRMarker的性能略有下降,这意味着在不同的α设置下,miRMarker相对稳定。较小的学习率α可能导致更好的性能,这与先前的研究结果一致[28]。在本研究中选择了α = 0.2。
模块数 g 的设置影响了在疾病样本区分中集成策略的效果。在线附录图S2B(http://bib.oxfordjournals.org/)展示了miRMarker在不同g设置({1, 3, 5, 7})下的平均AUC值。随着 g 从 1 变化到 5,miRMarker的性能不断显著提高。然而,当 g 从 5 变化到 7 时,miRMarker的性能改变变得平稳,表明性能难以进一步提高。g = 5 适用于大多数情况,并在本研究中被采用。
结直肠癌数据集上的生物标志物识别
我们在来自GEO数据库(GSE108153)的真实转录组学数据集上进行了miRMarker,以识别结直肠癌诊断的潜在miRNA生物标志物。结直肠癌是一种全球性的恶性肿瘤,面临着诸多危险因素,如吸烟、不健康的饮食和肥胖[38]。目前用于结直肠癌的治疗方法,包括放疗和切除术,对治愈率和预后的影响有限。对结直肠癌的诊断和预后的改进仍然是一个具有挑战性和吸引力的问题。
- 发现集(GSE108153)包含了来自结直肠癌患者的21对癌症和相邻正常组织样本。
- 从GEO数据库检索了涉及多种实体癌症的三个癌症转录组学数据集(GSE112264、GSE113486、GSE211692)。
- 对于每个数据集,我们提取了结直肠癌样本和非癌样本作为独立的验证集。在线附录表S5(http://bib.oxfordjournals.org/)提供了这三个独立验证集的详细信息。
- miRMarker在发现集上识别的潜在生物标志物在独立验证集上进行了验证。
miRMarker在发现集上定义了五个miRNA模块(图3A–E)。每个识别到的模块都是一个星形图。它们在区分癌症样本和正常样本方面表现出良好的性能(图3F)。
图4显示了miRMarker在发现集和三个独立验证集上的ROC曲线。在区分结直肠癌样本和正常样本时,miRMarker在发现集上获得了1.000的AUC。在三个独立验证集上,AUC值分别为1.000、0.9995和0.996。在发现集和独立验证集上,定义的模块在癌症样本区分方面表现出色,显示了作为结直肠癌生物标志物的巨大潜力。
此外,我们通过构建模块-疾病网络(图5)来探索定义的miRNA模块与结直肠癌之间的关系。所有五个定义的模块都与结直肠肿瘤的疾病节点相连接,表明这些定义的模块与结直肠肿瘤的发生和发展密切相关。有关模块-疾病网络的更多详细信息,请参阅在线附录材料
在生物网络中,高度连接的中心节点通常位于关键位置,对生物过程起着重要作用[39]。我们利用在线工具DAVID [40, 41]对五个中心miRNA(hsa-miR-135b-5p、hsa-miR-224-5p、hsa-miR-183-5p、hsa-miR-96-5p和hsa-miR-195-5p)的靶基因进行了通路分析。图6显示了Kyoto Encyclopedia of Genes and Genomes(KEGG)[42]的前30个富集通路。大多数顶部富集通路与细胞生长和衰老、信号传导、病毒或细菌感染以及与癌症相关的代谢有关。通常情况下,在KEGG通路“结直肠癌”(hsa05210)中富集了51个靶基因,虚发现率(FDR)为2.55e−06(在线附录表S6,http://bib.oxfordjournals.org/),表明受调控靶基因在结直肠癌的发生和发展中发挥着重要作用。
为了进一步了解miRNA在结直肠癌发展中调控基因的机制,我们使用了癌症基因组图谱(TCGA)结直肠癌队列进行基因表达分析。结直肠癌的TCGA基因表达数据来自UCSC Xena平台(http://xena.ucsc.edu/)。基因表达分析包括了41个正常样本和453个原发性结直肠肿瘤样本。在线附录图S3(http://bib.oxfordjournals.org/)显示了‘结直肠癌’(hsa05210)这一通路的异常调控概览。在结直肠癌中,靶基因的失调涉及到重要的细胞功能通路‘细胞周期’(hsa04110,富集FDR=5.01e−12)、‘凋亡’(hsa04210,富集FDR=3.75e−03)以及包括Wnt信号通路(hsa04310,富集FDR=2.20e−04)、MAPK信号通路(hsa04010,富集FDR=3.78e−05)和p53信号通路(hsa04115,富集FDR=2.36e−05)等几个关键信号通路。可以看出,一个中心miRNA的失调会导致多个与结直肠癌相关的通路的干扰,共同促进结直肠癌的发展。有关通路中基因表达分析的更多详细信息,请参阅在线附录材料(http://bib.oxfordjournals.org/)。
癌症的发展涉及多种生物过程。不同的通路相互作用,并共同影响肿瘤过程。miRNA与靶基因之间存在复杂的调控关系。实验结果显示,所识别的miRNA模块不仅在疾病样本区分方面具有出色的能力,而且通过参与多个相关通路的扰动,在结直肠癌的发生和发展中发挥着关键作用。所识别的miRNA有望成为临床应用中潜在的结直肠癌生物标志物。需要进行更多实验证实所识别的miRNA在结直肠癌中的生物学意义。
其他疾病的实验
此外,我们还进行了另外三个实验,以进一步验证miRMarker的有效性。考虑了三个具有挑战性的任务:鼻咽癌(NPC)的预后、复发性植入失败(RIF)的诊断以及COVID-19的严重判断。在线附录表S7(http://bib.oxfordjournals.org/)提供了涉及的发现集和独立验证集的详细信息。
与对结直肠癌的操作一样,miRMarker在发现集上识别的潜在生物标志物在相应的独立验证集上得到了验证。在线附录表S8(http://bib.oxfordjournals.org/)显示了这三个实验的结果。对于NPC实验,miRMarker在发现集上获得了0.922的AUC,在独立验证集上获得了0.731的AUC。对于RIF实验,miRMarker在发现集和独立验证集上的AUC值分别为0.989和0.967。对于COVID-19实验,miRMarker在发现集上获得了0.895的AUC,在独立验证集上获得了1.000的AUC。
这三个实验的结果进一步说明了miRMarker在识别不同疾病的潜在生物标志物方面的有效性。有关这三个实验的更多详细信息,请参阅在线附录材料(http://bib.oxfordjournals.org/)。总之,miRMarker在疾病生物标志物识别方面展现出其优势。
结论
识别用于疾病(如癌症)诊断和预后的信息丰富的miRNA具有吸引力且具有挑战性。基于网络的数据分析方法是潜在生物标志物识别中的重要工具。
在这项研究中,我们提出了一种基于多视图网络和强化学习的miRNA数据分析方法miRMarker,用于定义疾病的潜在miRNA生物标志物。
miRMarker分别基于miRNA表达数据和已知miRNA-疾病关系构建合作调控网络和功能相似性网络。然后,采用强化学习策略来定义区分不同样本组的潜在miRNA模块生物标志物。
我们在多个miRNA数据集上将miRMarker与八种高效的数据分析方法(SVM-RFE、WGCNA、DNB、GroupBN、NGTM、QLCD、N-DAG和DDRM)进行了比较。miRMarker在疾病样本区分方面表现出更好的性能,表明它基于miRNA表达数据和知识库识别了有价值的miRNAs。
此外,我们将miRMarker应用于定义结直肠癌的潜在生物标志物。所定义的miRNA模块通过调控多个相关通路在结直肠癌的发展中起着关键作用。实验结果说明miRMarker识别的miRNAs在疾病样本区分方面具有出色的能力,并且在结直肠癌的发展中具有重要的生物学意义。整合多视图miRNA网络有助于构建稳健的miRNA相互作用网络。
此外,通过强化学习定义miRNA模块可以充分探索网络信息,并识别更具竞争力的miRNA生物标志物,用于疾病样本区分。总的来说,miRMarker在人类疾病的miRNA数据分析中具有很大的潜力。
需要承认这项工作存在一些局限性。miRNA功能相似性网络依赖于公共知识库中已知的miRNA-疾病关系。进一步更新和丰富公共知识库将有助于评估miRNA功能相似性。同时,构建的miRNA功能相似性网络被设计成适用于不同疾病,这可能未能突显某些疾病的特定特征。一个可能的改进是考虑构建特定疾病的特定功能相互作用网络。此外,在未来的工作中,我们计划开发miRMarker的并行版本,以进一步提高疾病生物标志物识别的效率。
要点
• miRNA相互作用网络是整合基于表达数据和已知miRNA-疾病关系的多视图miRNA网络构建的。
• 在计算基于已知miRNA-疾病关系的miRNA功能相似性时采用了自然语言处理。
• 使用强化学习策略定义了与疾病发展相关的重要miRNA模块。
• 与八种数据分析方法的比较结果说明了miRMarker的有效性。
• 所定义的模块不仅在疾病样本区分方面具有出色的能力,且在相关通路的扰动中起着关键作用。


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









