






 本文详细介绍了代谢组学的研究对象(尿液、血浆等)、检测能力(小分子代谢组学和脂质组学),以及实验设计、数据处理(基峰图与总离子流图的区别)、数据分析方法(如PLS-DA和OPLS-DA)和差异代谢物筛选。还探讨了为何代谢组学结果不同于蛋白组学,以及差异代谢物验证的方法。
本文详细介绍了代谢组学的研究对象(尿液、血浆等)、检测能力(小分子代谢组学和脂质组学),以及实验设计、数据处理(基峰图与总离子流图的区别)、数据分析方法(如PLS-DA和OPLS-DA)和差异代谢物筛选。还探讨了为何代谢组学结果不同于蛋白组学,以及差异代谢物验证的方法。
代谢组学可以检测的样品类型
代谢组学主要研究对象是各种代谢小分子产物(MW<1000)及脂质。其样品主要是尿液,血浆或血清,脑脊液,唾液,以及细胞,组织,粪便及肠道内容物。当然,植物,真菌,微生物,土壤,以及比较特殊的样本如淋巴液,羊水,卵泡液,膝盖滑液,眼泪,精液等,都可以作为代谢组学的检测对象。
代谢组学的检测能力
非靶向代谢组学,主要包括针对小分子代谢组学和脂质组学。
小分子代谢组学
我们建立了约2700多种代谢物的LC-MS/MS本地标准品数据库。通过LC-MS/MS技术分析平台检测到的代谢物都必须与标准品库中的代谢物进行一级和二级的匹配。这些标准品库数据包括氨基酸及其衍生物、核苷酸及其衍生物、糖代谢途径中的各种代谢物、生物碱、黄酮类、萜类、酚胺类、激素及其衍生物、脂肪酸类等。
不同物种、不同组织检测到的代谢物不一样,一般针对细胞样本,通过正负离子检测,大概能定性到300个左右的物质。很多客户会问为什么不用HMDB,METLIN等公共库查库,这是因为检索公共库只能通过一级谱库来定性,在检索的过程中缺少二级支持(部分物质有二级信息,但由于仪器和设置方法等不一样,匹配结果可靠性较差),这样会产生大量假阳性鉴定结果,后期分析非常繁琐。成熟代谢组学分析实验室基本上都在自己建立数据库。
脂质组学
我们利用的是Thermo Scientific™ 公司研发的LipidSearch™ 软件(Thermo Scientific™)进行脂质鉴定与数据预处理。LipidSearch™ 软件能够实现原始数据处理、峰提取、脂质鉴定、峰对齐、和定量等的一体化分析。LipidSearch™中收录了8大类,300种亚类,约170 万种脂质分子的MS2 & MS3数据库,并基于Orbitrap™质谱仪生成的高分辨率高质量精度数据,通过子离子、母离子和中性丢失扫描的鉴别算法,实现系统地、可靠地脂质定性分析。一般细胞的脂质组项目,我们大概能鉴定到1000个左右的脂质。
代谢组学实验设计
如果是转录组学或蛋白组学,一般一组样本有3-4次生物学重复就足够了,如果一组有多个样本,可以进行随机混合并形成3个生物学重复,但是代谢组学却不是这样。
由于个体的代谢谱差异较大,我们不建议混样,一般建议植物,微生物设计6-8个平行样品,模式动物设计10个左右平行样品,临床标本设计20-30例平行样品(所有样品当然越多越好)。注意,样品不能pooling,不能反复冻融,血清/血浆不能溶血。另外,如果是用血浆,请用肝素钠抗凝。
基峰图(BPC)和总离子流图(TIC)有什么区别
基峰图(Base Peak Chromatogram,BPC):是将每个时间点质谱图中最强的离子的强度连续描绘得到的图谱。
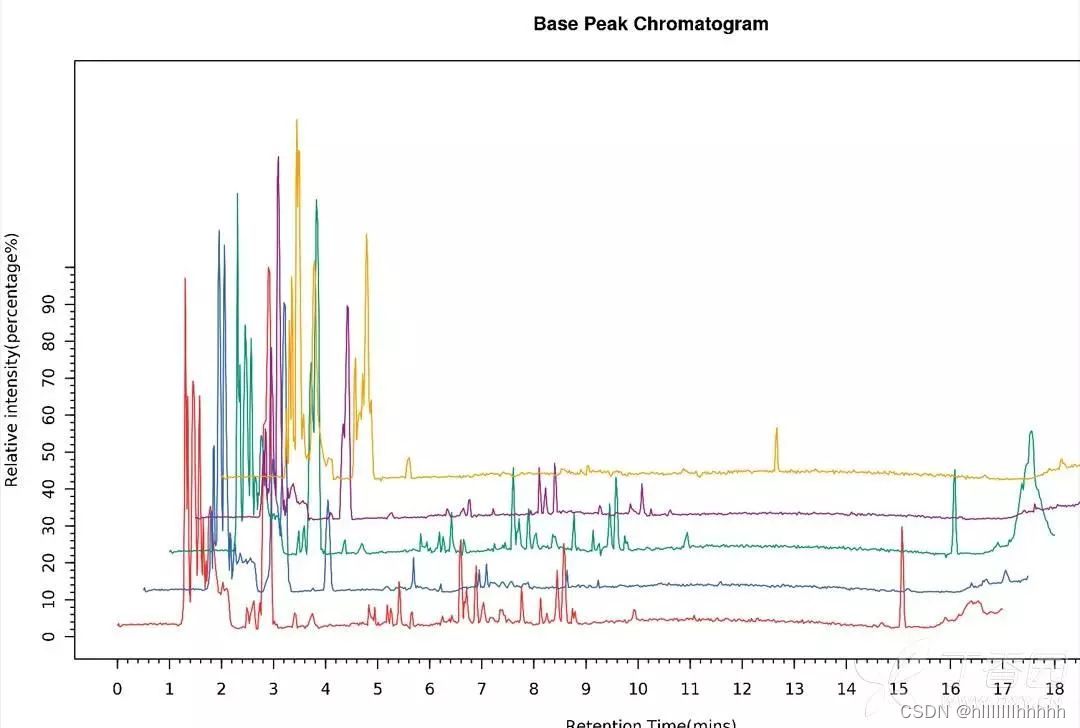
总离子流图(TIC):在选定的质量范围内,所有离子强度的总和对时间或扫描次数所作的图。
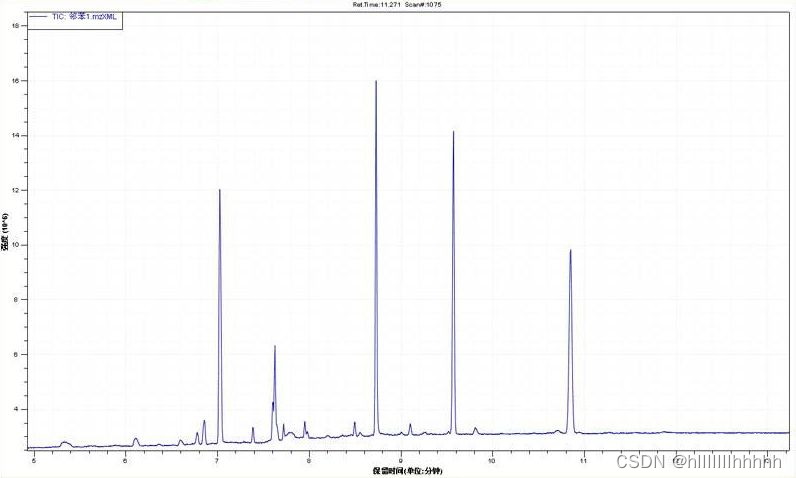
TIC和BPC都是对于样品整体信息的反映,一般情况下BPC图比TIC图要漂亮,所以文章里面很多时候会用到BPC图。但是有的学者认为BPC图不是样品真实的反映,所以不接受BPC,只接受TIC。
代谢组学数据分析
数据分析的内容包括:
step1:XCMS数据预处理
step2:数据多位统计学分析:PCA分析;PLS-DA分析;OPLS-DA分析;permutation test分析等
step3:差异化合物筛选
step4:差异化合物鉴定
step5:差异化合物聚类分析和关联分析
step6:代谢通路分析和富集分析
step7:多组学数据关联分析(具备多组学数据)
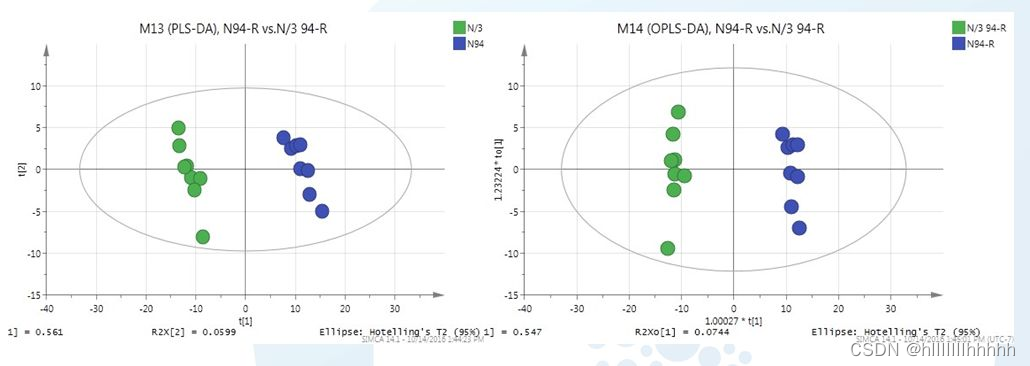
PLS-DA和OPLS-DA模型有什么区别
OPLS-DA比PLS-DA多了一个正交换算,把与模型分类不相干的信号过滤掉,因此OPLS-DA解释能力更强。比如组间差异比较小,组内差异比较大时,用PLS-DA的VIP筛选出的可能是组内差异变量,容易误导,而OPLS-DA则优于PLS-DA,可更准确地筛选出组间差异。
为何代谢组学结果不像蛋白质组学结果提供所有鉴定的化合物
- 一般文献不体现鉴定到总的代谢物,而是描述实验总共检测到多少离子峰,进行差异分析后鉴定到多少个代谢物。 因代谢物存在多种同分异构现象较多,且存在加和形式的多样性,如果定性结果没有经过核验,会存在很多冗余甚至假阳性的结果,所以一般不建议直接使用。
- 代谢组学一般先定量后定性,即先通过多维统计和单变量统计学方法寻找有差异的离子,后经过查库比对,人工校验得到准确的代谢物名称及结构,这样是常用的思路和分析方法。所以不提供所有鉴定的化合物。
差异代谢物的筛选标准
- 一般同时采用多元变量统计模型的VIP值和单变量统计T检测的P值来筛选差异代谢物。
- 单变量统计分析方法如t检验、方差分析等更加注重代谢物水平的独立变化
- 多元变量统计分析更加注重代谢物之间的关系以及它们在生物过程中的促进/拮抗关系。同时考量两类统计分析方法的结果,有助于我们从不同角度观察数据,得出结论,也可以帮助我们避免只使用一类统计分析方法带来的假阳性错误或模型过拟合。
- 筛选的阈值一般是VIP>1和P<0.05。如果得到的差异代谢物很多,可以再加上差异倍数这个筛选条件。
筛选不到差异代谢物怎么办
- 如果利用常用的筛选标准(VIP>1和P<0.05)没有找到差异代谢物,首先可以筛选阈值设得宽放些,如VIP>1和P<0. 1。
- 如果还是没有筛选出差异代谢物,那么还可以对检测到的物质进行单维统计学分析,如FC>1.5和P<0.05,通过每单个代谢物组间样本的表达值比较分析差异代谢物,并用火山图展现。
找到差异代谢物之后,如何验证这个代谢物的功能
- 首先要进行定性定量验证。主要是采用三重四级杆进行靶向验证,获得绝对定量信息。
- 然后通过前期的代谢通路分析,我们知道这个差异代谢物属于哪条调控通路,那么可以对该通路上的分子进行功能验证。
- 另外,还可以增加其他组学的数据,如转录组、蛋白组,进行相互补充。

















 3759
3759

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









