



通常使用第一性原理计算某些多元素占据原胞中同一位置的结构会优先考虑使用准随机结构(special quasirandom structure,SQS)来进行模拟建模。此篇教程意在整理一个较为简便的操作流程,以供参考。
合金理论自动化工具包(ATAT)1是一个通用名称,指的是合金理论的集合 Axel van de Walle2与多个研究小组合作开发的工具。而由于高熵合金内部多元、混合等特性,ATAT工具包内的mcsqs模块所涉及的特殊准随机结构Special quasi-random structure(SQS)于此有了非常大的应用需求。
部分有关mcsqs模块的使用教程如下
1、建无序格点参杂&合金模形工具包ATAT的安装与其各模块的详细使用例子
3、高熵合金的mcsqs建模(https://www.bilibili.com/video/BV1QK4y1h752?spm_id_from=333.337.search-card.all.click&vd_source=4a437e68a7f05cd2b6b8232814f71b42)
首先,准备原始单一元素(共格的多元素合并为一种)晶胞,并通过vaspkit获得其最原始晶胞。
例如这里准备了一个面心立方的结构,组分单一,面心和顶角元素相同,这里设置为Si。
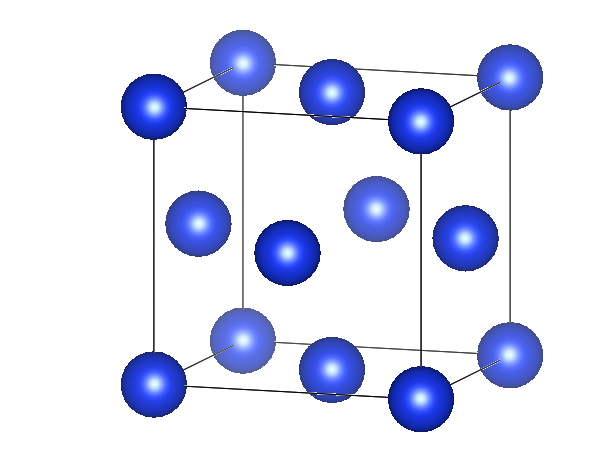
Si1.000000000000004 0.0000000000000000 0.00000000000000000.0000000000000000 4 0.00000000000000000.0000000000000000 0.0000000000000000 4Si4Direct0.0000000000000000 0.0000000000000000 0.00000000000000000.0000000000000000 0.5000000000000000 0.50000000000000000.5000000000000000 0.0000000000000000 0.50000000000000000.5000000000000000 0.5000000000000000 0.0000000000000000
通过vaspkit 602功能获得其原胞
vaspkit -task 602+---------------------------------------------------------------+| VASPKIT Standard Edition 1.3.5 (03 Jul. 2022) || Running VASPKIT Under Command-Line Mode |+---------------------------------------------------------------+-->> (01) Reading Structural Parameters from POSCAR File...+-------------------------- Summary ----------------------------+Prototype: ATotal Atoms in Input Cell: 4Lattice Constants in Input Cell: 4.000 4.000 4.000Lattice Angles in Input Cell: 90.000 90.000 90.000Total Atoms in Primitive Cell: 1Lattice Constants in Primitive Cell: 2.828 2.828 2.828Lattice Angles in Primitive Cell: 60.000 60.000 60.000Crystal System: CubicCrystal Class: m-3mBravais Lattice: cFSpace Group: 225Point Group: 32 [ Oh ]International: Fm-3mSymmetry Operations: 192+---------------------------------------------------------------+-->> (02) Written PRIMCELL.vasp file.+---------------------------------------------------------------+
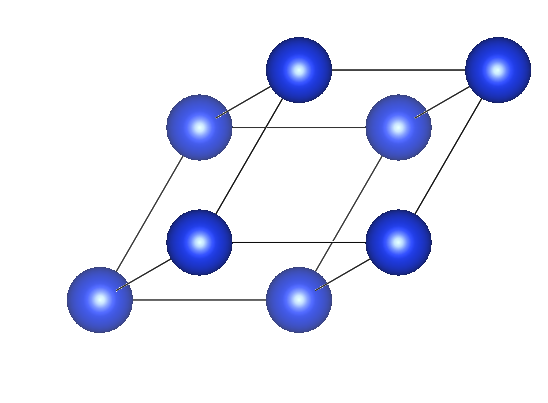
原胞信息如下,只存在一个wyckoff点位。
Primitive Cell1.0000000.00000000000000 2.00000000000000 2.000000000000002.00000000000000 0.00000000000000 2.000000000000002.00000000000000 2.00000000000000 0.00000000000000Si1DIRECT0.0000000000000000 0.0000000000000000 0.0000000000000000 Si1
将vaspkit生成的原胞文件PRIMCELL.vasp复制为POSCAR,然后通过vaspkit 的414功能(VASPKIT版本需低于1.5)将POSCAR转化为ATAT的输入文件lat.in
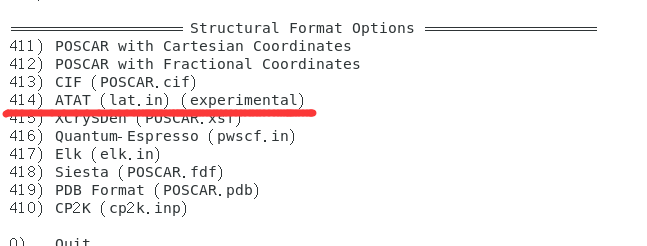
vaspkit -task 414+---------------------------------------------------------------+| VASPKIT Standard Edition 1.3.5 (03 Jul. 2022) || Running VASPKIT Under Command-Line Mode |+---------------------------------------------------------------+-->> (01) Reading Structural Parameters from POSCAR File...-->> (02) Written lat.in File!+---------------------------------------------------------------+
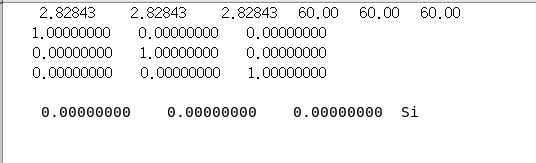
lat.in文件内容如下,后续计算需要重命名为rndstr.in
根据实际需要的某一wyckoff点位的元素比例修改rndstr.in文件。这里修改为Si和C各占50%。修改后的rndstr.in文件如下
2.82843 2.82843 2.82843 60.00 60.00 60.001.00000000 0.00000000 0.000000000.00000000 1.00000000 0.000000000.00000000 0.00000000 1.000000000.00000000 0.00000000 0.00000000 Si=0.5,C=0.5
注意:各点位各元素之间的比例与扩包比例相关联,最后确保扩包后各元素原子数量总数为整数。
因VASPKIT版本问题,新版本不再支持转换lat.in文件,建议使用ATOMKIT进行格式转换。
注:笔者关注到目前众多使用者的需求是获得晶格矢量正交的惯用胞超胞,在此特意更新注明,有两个途径来达成此需求。
一、跳过刚才的一步获得原胞的过程,直接基于惯用晶胞来生成rndstr.in文件。
4 4 4 90 90 901.00000000 0.00000000 0.000000000.00000000 1.00000000 0.000000000.00000000 0.00000000 1.000000000.0000000000000000 0.0000000000000000 0.0000000000000000 Si=0.5,C=0.50.0000000000000000 0.5000000000000000 0.5000000000000000 Si=0.5,C=0.50.5000000000000000 0.0000000000000000 0.5000000000000000 Si=0.5,C=0.50.5000000000000000 0.5000000000000000 0.0000000000000000 Si=0.5,C=0.5
在后续计算过程中,会提示非最基本原胞的警告,但不影响计算和生成结构。
二、更改sqscell.out文件(请先阅读后续流程,熟悉晶格矢量变化后自行换算)
这里选择扩包方式为2×2×2,最后超胞总原子数量为8。
先使用ATAT获得in文件的对称性信息,-2=x需要根据结构实际情况修改,过小会导致ATAT出现段错误。
corrdump -nop -noe -2=4 -ro -l=rndstr.in -clus ; getclus再根据超胞的总原子数获得结构信息,-n=后面的数值与超胞数量相同
mcsqs -n=8ATAT产生的超胞结构信息文件为sqscell.out文件
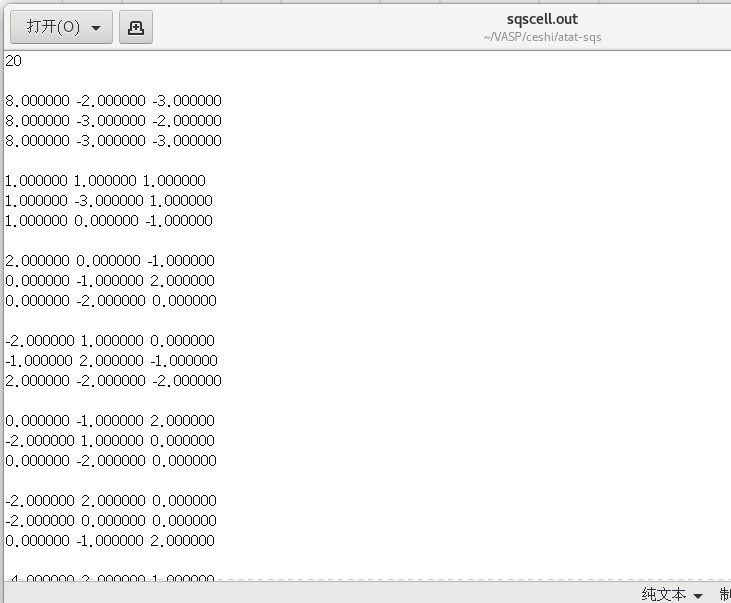
根据既定扩包方式修改为:
12 0 00 2 00 0 2
生成超胞准随机结构:
mcsqs -rc产生的准随机结构文件为:bestsqs.out
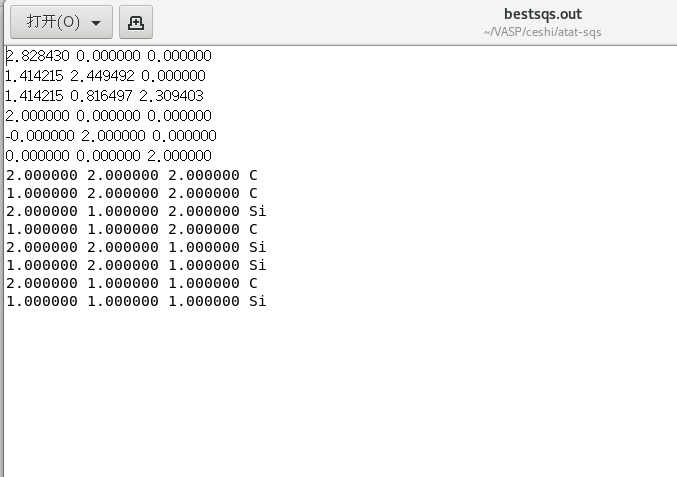
导出为cif文件
str2cif < bestsqs.out > pos.cif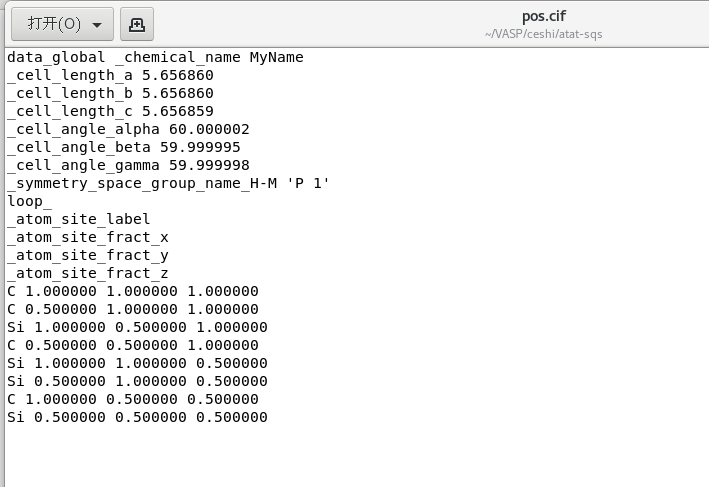
推荐:使用VESTA进行cif向POSCAR的转换.
通过vaspkit 将cif文件转化为POSCAR
vaspkit -task 105+---------------------------------------------------------------+| VASPKIT Standard Edition 1.3.5 (03 Jul. 2022) || Running VASPKIT Under Command-Line Mode |+---------------------------------------------------------------+Input the filename in cif format (e.g., POSCAR.cif):------------>>pos.cif-->> (01) Written POSCAR file!
更为简便的方法可使用qvasp软件一键转化
qvasp -c2p当然也可以通过VESTA导出。
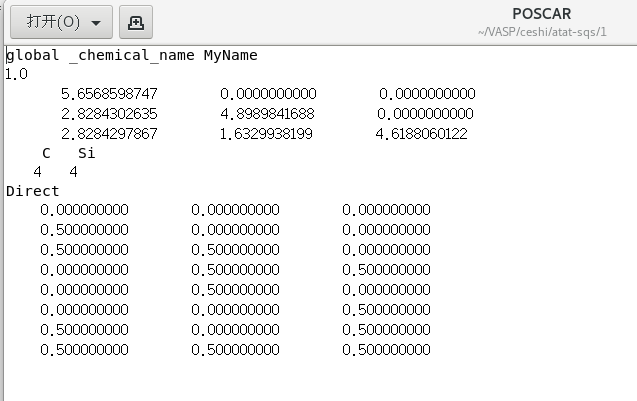
POSCAR文件如下


















 7023
7023

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









