







原文标题:Molecular design in drug discovery: a comprehensive review of deep generative models
基于smiles的模型和基于图的模型的综述。
一、分子表征(SMILES & Graph)
SMILES的缺点:(1)不能捕捉分子结构的相似性。两个相似结构之间的微小变化可能会导致SMILES字符串有很大的不同,从生成模型中学习到的潜在空间并不平滑。(2) SMILES字符串是非唯一的,一个分子可以被编码成多个SMILES表示。
数据库:其中ZINC、ChEMBL、PubChem和DrugBank使用最多。
二、Deep molecular generative models
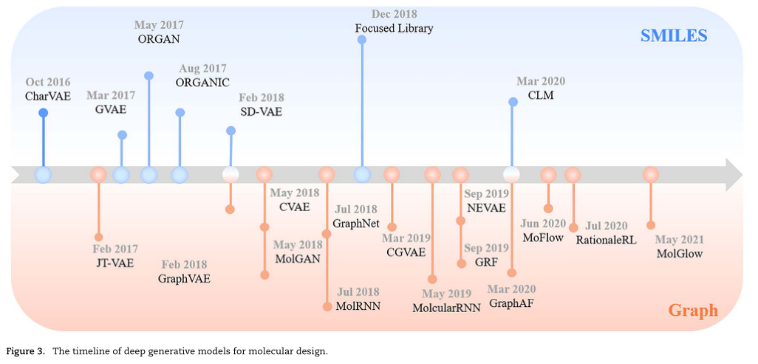
1、SMILES-based Model
基于序列的方法可以进一步分为基于变分编码器(VAE)、生成对抗网络(GANs)和基于循环神经网络(RNNs)的模型。
VAE-based generative models
VAE分子生成的潜在空间是潜在操作的,如控制特定性质,训练过程是稳定的。然而,训练集的重构限制了在未知化学空间中的探索能力。在没有额外约束的情况下,VAE模型产生无效分子的概率很高
【Automatic chemical design using a data-driven continuous representation of molecules】:使用核密度估计来学习捕获分子的相关特征。然后在维度上学习连续的潜在空间,优化分子的特定性质,允许使用强大的基于梯度的方法来有效地指导搜索。加入多层感知器和编码器的联合训练任务,保证了分子性质的预测能力。
【Grammar variational autoencoder】:将SMILES的语法生成规则纳入模型。它表明可以使用上下文无关语法将离散数据直接表示为解析树。解码器通过按顺序学习这些规则来生成有效的输出。考虑到解析树,该模型可以扩展到其他没有上下文的文本表示学习。
【Syntax-directed variational autoencoder for molecule generation】:提出GVAE缺乏语义,生成的环键必须紧密等结构信息。然而,在GVAE中增加额外的结构约束可能会造成不必要的计算和时间浪费。
GANs-based generative models
【latentGAN:A de novo molecular generation method using latent vector based generative adversarial network】:autoencoder and GAN的结合。
RNNs-based generative models
语言模型从语法和语义两方面自动提取信息。由于SMILES的序列表示,自然语言处理任务与分子生成的类比是可行的
【Generating focused molecule libraries for drug discovery with recurrent neural networks.】:通过重新训练模型生成聚焦分子库(见图2.3)。大规模数据集的采样确保了分子的多样性,微调增加了聚焦特性。
【QBMG: quasi-biogenic molecule generator with deep recurrent neural network】:建立了包含立体化学性质的准生物源化合物库。除了添加迁移学习外,利用计算模型CLM ,结合之前的三种优化方法(数据增强、温度采样和迁移学习),在化学空间的指定区域设计新分子。
【Generative molecular design in low data regimes】:分子描述符被纳入基于rnn的模型,这比传统方法更有针对性。
【Randomized smiles strings improve the quality of molecular generative models】:用20亿次替换的方法进行了研究,并探索了GDB-13中SMILES符号的三种不同变体(规范的、随机的和深度的)来证明‘与随机化SMILES相比,规范SMILES形式创建有效和语义结构的大型化学空间的能力较低。’
【SMILES-based deep generative scaffold decorator for de-novo drug design】:利用切片算法获得具有随机化SMILES表示的支架集。然后部分构建的分子被装饰在一个附着物上一次或多次
基于smiles的模型激增,但仍然存在一些亟待解决的问题:不仅面临着有效性的问题,而且SMILES的非结构化性质使得两个相似的分子在很大程度上有很大的不同。将有效性约束强制纳入解码器是昂贵的,这需要设计具有更多结构信息的新表示。
2、Graph-based models
VAE-based generative models
最具代表性的是junction tree variational autoencoder, JT -VAE:通过分解训练集的分子,得到了环、官能团和原子等子结构。与之前逐节点生成图相比,整个过程分为两个阶段,首先将有效支架及其排列表示为树,然后通过在交叉组件之间添加边将整个树集成到图中。
这种设计有三个关键的局限性。首先,由于具有相同junction tree的两个分子可能对应着明显不同的属性,所以使用JT VAE进行性能优化更加困难。其次,在生成过程中不考虑节点的顺序排列,造成耗时。在一些可能的节点排列下的最终序列可以映射到同一个图中。第三,由于现实中药物分子的复杂性,一个子结构中小于20个原子是不现实的。
【Learning multimodal graph-to-graph translation for molecule optimization.】分子优化任务视为图到图的转换,目的是学习两个域之间的多模型映射。
将多种属性整合到一个分子中是一个挑战,原因有两个:(1)在现实中缺乏符合所有约束条件的分子,如效力、安全性和期望的属性;(2)在添加所有属性约束条件时,成功率和新颖性表现不佳。
【Constrained graph variational autoencoders for molecule design,CGVAE】纳入了两种类型的相关性,包括一些已知的规则,如价规则为硬规则,环应变(不利于循环)为软规则。保持CGVAE的相关性,将其转化为语义上有效的SMILES。值得注意的是,具有相似性质的分子实际上在节点和边的大小上存在差异,而GraphVAE等分子图生成模型并没有考虑到这一点。
【NEVAE: A deep generative model for molecular graphs.】提出了不同跳数的neae聚合特征。同时,neae生成的图在节点层面具有排列不变性,并考虑了空间位置对属性的影响。
GANs-based generative models
【Mol-CycleGAN: a generative model for molecular optimization】:数据集分为两部分,一部分不配备目标属性(即卤素基团,芳香族环数,活性)用于训练,另一部分则相反,用于测试。框架为:(1)数据集:集合X(例如,CN组)和Y(例如,CF3组);(2)映射:G: X→Y和F: Y→X)周期一致性损失:鼓励F(G(X))≈X和G(F(Y))≈Y)指标:给定属性的优化值,如logP。为缺乏配对样本的实际情况下的分子优化提供了新的思路。受MolCycleGAN的启发,可以将分子优化视为机器翻译或图翻译的问题。
RNNs-based generative models
【Learning deep generative models of graphs,GraphNet】是第一个基于rnn的任意图模型,它建立在消息传递神经网络(MPNN)的框架上,在已有的图中添加一个新的原子或键。更具体地说,(1)选择是否添加原子,(2)计算现有图上的概率,以确定是否添加新边,(3)计算图中某个节点连接的概率。
【Multi-objective de novo drug design with conditional graph generative model】:探索了基于图卷积网络(graph convolutional networks, GCN)的MolMP和MolRNN与GraphNet的生成类似,都是通过在已有的子图上迭代地添加节点和边来生成分子。将额外的约束转换为不需要强化学习的可用条件代码提供了更高的灵活性,并输出了更具多样性的分子。
【Generating realistic graphs with deep auto-regressive models,GraphRNN】是一种基于图和边层的层次模型,旨在捕获节点和边的联合概率。图的生成过程被看作是不同节点顺序下邻接向量的序列。通过引入宽度优先搜索(BFS)节点排序来具备可伸缩性。随后,从GraphRNN扩展了一些分子生成的工作。
【MolecularRNN: Generating realistic molecular graphs with optimized properties 】:在GraphRNN的基础上,添加与其相关的节点和边特征向量。模型中插入基于价格的剔除抽样,以确保有效分子率100%。它体现了大数据集预训练与策略梯度算法调优相结合的特点和优势。借助于强化学习,在惩罚logP系数和QED方面优于JT VAE、GCPN和ORGAN。
Flow-based generative models
基于流的生成模型借助流的归一化,明确地学习由可逆变换组成的数据分布。该流以初始变量为输入,通过反复使用变量变换规则将其转换为具有各向同性高斯分布的变量,这类似于VAE编码器中的推理过程。
现有的基于图的模型大致可以分为两种类型,一种是顺序迭代过程,另一种是一次性生成。具体地说,它们可以划分为逐个原子、基于子图(片段)的模型。
【GraphNVP: An invertible flow model for generating molecular graphs】:第一个基于流的分子图生成模型,提高了分子的唯一性。
【Graph residual flow for molecular graph generation,GRF】:在减少参数数量的情况下,性能几乎相当。不幸的是,GraphNVP和GRF模型在生成有效分子方面表现不佳。
【GraphAF: a flow-based autoregressive model for molecular graph generation】:受自回归和少数基于流的模型的启发,提出了一种基于流的自回归序列模型,优于当代最先进的模型图卷积策略网络,并通过结合价性检查生成100%有效分子。
【MoFlow: an invertible flow model for generating molecular graphs】通过一种新的图条件流,通过Glow的变体和具有给定键的原子生成键,提出了一种新的有效性修正方法,即递归地删除上一阶键,保持最大有效分量。
【MolGrow: A graph normalizing flow for hierarchical molecular generation.】利用模型的潜在变量进行了性能约束优化,递归地将一个节点分成两个,生成分子结构,可以看作是即插即用模块。
从现在来看,论文中的方法都是比较老的方法,大部分是2020年之前的方法。但是参考性相对较大。更多最新的文章可以查看最新的分子生成优化综述:Drug Discov. Today2022 | Deep learning methods for molecular representation and property prediction_羊飘的博客-CSDN博客_smiles编码转换到onehot


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









