







背景介绍
单细胞 RNA-seq(scRNA-seq)技术已经成为解析细胞异质性和基因表达调控的重要工具。目前最常见的 scRNA-seq 平台是 10x Genomics,其官方提供的分析工具 Cell Ranger 计算资源消耗较大,尤其在处理大规模数据时,计算瓶颈明显。为了解决这一问题,STARsolo 作为一种更轻量、高效的替代方案,提供了灵活的单细胞转录组数据处理方法。
STARsolo 是基于 STAR(Spliced Transcripts Alignment to a Reference) 的扩展模块,专门用于 10x Genomics、Drop-seq、inDrop、BD Rhapsody、BGI STOmics 等单细胞测序平台的数据分析。
本文将介绍 STARsolo 的优势、使用方法、关键参数设置及不同平台数据的分析流程。
1. 为什么选择 STARsolo?
1.1 STARsolo vs. Cell Ranger 对比
| 对比项 | STARsolo | Cell Ranger |
|---|---|---|
| 比对工具 | STAR | 内置比对 |
| 运行速度 | 更快(更少的内存占用) | 计算资源消耗大 |
| 支持平台 | 10x, Drop-seq, inDrop, BD, BGI | 仅 10x Genomics |
| 灵活性 | 可调整参数,更灵活 | 封闭,依赖 10x 生态 |
| 是否支持 TCR/BCR 组装 | 不支持 | 支持 |
| 是否支持 Feature Barcode | 支持 | 支持 |
1.2 适用场景
- 数据量大,Cell Ranger 计算资源消耗过高
- 需要支持多种测序平台(Drop-seq, BD, BGI)
- 希望自定义比对和 UMI 处理流程
2. STARsolo 运行流程
2.1 处理 10X Genomics v3 数据
STAR --runThreadN 16 \
--genomeDir /path/to/genome \
--readFilesIn sample_R2.fastq.gz sample_R1.fastq.gz \
--readFilesCommand zcat \
--soloType CB_UMI_Simple \
--soloCBwhitelist /path/to/10x_v3_whitelist.txt \
--soloBarcodeReadLength 16 \
--soloUMIlen 12 \
--soloFeatures Gene GeneFull \
--outFileNamePrefix star_output/
2.2 处理 Drop-seq 数据
STAR --runThreadN 16 \
--genomeDir /path/to/genome \
--readFilesIn sample_R2.fastq.gz sample_R1.fastq.gz \
--readFilesCommand zcat \
--soloType Droplet \
--soloCBstart 1 --soloCBlength 12 \
--soloUMIstart 13 --soloUMIlength 8 \
--outFileNamePrefix star_output/
2.3 处理 BD Rhapsody 数据
STAR --runThreadN 16 \
--genomeDir /path/to/genome \
--readFilesIn sample_R2.fastq.gz sample_R1.fastq.gz \
--readFilesCommand zcat \
--soloType CB_UMI_Complex \
--soloCBmatchWLtype 1MM_multi_Nbase_pseudocounts \
--soloBarcodeReadLength 15 \
--soloUMIlen 8 \
--soloFeatures GeneFull Gene \
--outFileNamePrefix star_output/
3. 关键参数解析
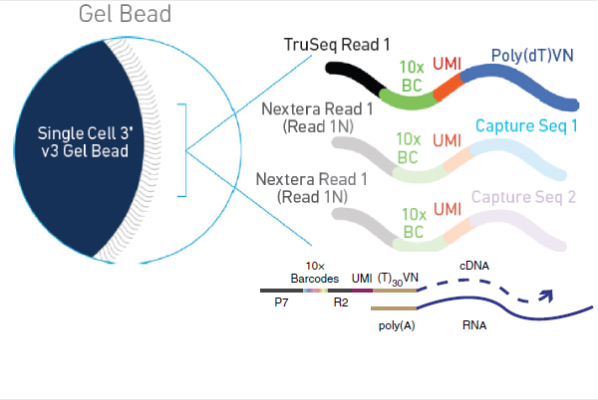
3.1 --soloBarcodeReadLength:条形码(Cell Barcode, CB)长度
Cell Barcode(CB) 是单细胞测序时用于标记不同细胞的唯一标签,通常位于 Read 1(R1)。
| 测序平台 | 条形码位置 | 条形码长度(bp) |
|---|---|---|
| 10X Genomics v2 (3’) | Read 1 | 16 |
| 10X Genomics v3 (3’) | Read 1 | 16 |
| 10X Genomics v3 (5’) | Read 1 | 16 |
| Drop-seq | Read 1 | 12 |
| inDrop | Read 1 | 8 |
| BD Rhapsody | Read 1 | 15 |
| BGI STOmics | Read 1 | 16 |
3.2--soloUMIlen:唯一分子标识符(UMI)长度
UMI(Unique Molecular Identifier) 是单细胞 RNA-seq 数据去重的关键。
由于 PCR 扩增会产生大量重复 reads,UMI 允许我们去除 PCR 复制,并计算真实的 RNA 分子数量。
UMI 的工作原理:
- 每个 RNA 分子在文库构建时被赋予一个随机的 UMI 标签。
- 经过 PCR 扩增后,相同 RNA 分子的 reads 可能被测序多次。
- 通过 UMI,可以识别和去重重复的 PCR reads,仅保留真实的原始 RNA 分子。
| 测序平台 | UMI 位置 | UMI 长度(bp) |
|---|---|---|
| 10X Genomics v2 (3’) | Read 1 | 10 |
| 10X Genomics v3 (3’) | Read 1 | 12 |
| 10X Genomics v3 (5’) | Read 1 | 12 |
| Drop-seq | Read 1 | 8 |
| inDrop | Read 1 | 6 |
| BD Rhapsody | Read 1 | 8 |
| BGI STOmics | Read 1 | 10 |
4. STARsolo 结果解析
运行完成后,star_output/ 目录包含:
star_output/
├── Solo.out/Gene/ # 细胞-基因表达矩阵
│ ├── barcodes.tsv # 细胞条形码
│ ├── genes.tsv # 基因名
│ ├── matrix.mtx # 稀疏矩阵
├── Log.final.out # 运行日志
├── Aligned.sortedByCoord.out.bam # BAM 文件
这些文件可直接用于 Seurat / Scanpy 进行后续分析。
library(Seurat)
data <- Read10X(data.dir = "star_output/Solo.out/Gene/")
seurat_obj <- CreateSeuratObject(counts = data)
5. 结论与推荐
- STARsolo 计算资源消耗低,适合处理大规模数据,是 Cell Ranger 的优秀替代方案。
- 支持多种测序平台(10x, Drop-seq, BD, BGI),比 Cell Ranger 更灵活。
- 不支持 V(D)J 组装(TCR/BCR 分析),如有此需求建议使用 Cell Ranger。
- 适用于云计算、自定义 pipeline 及大规模 scRNA-seq 数据分析。
如果在使用 STARsolo 处理数据时遇到问题,欢迎留言交流!



















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











