






https://github.com/mli/paper-reading
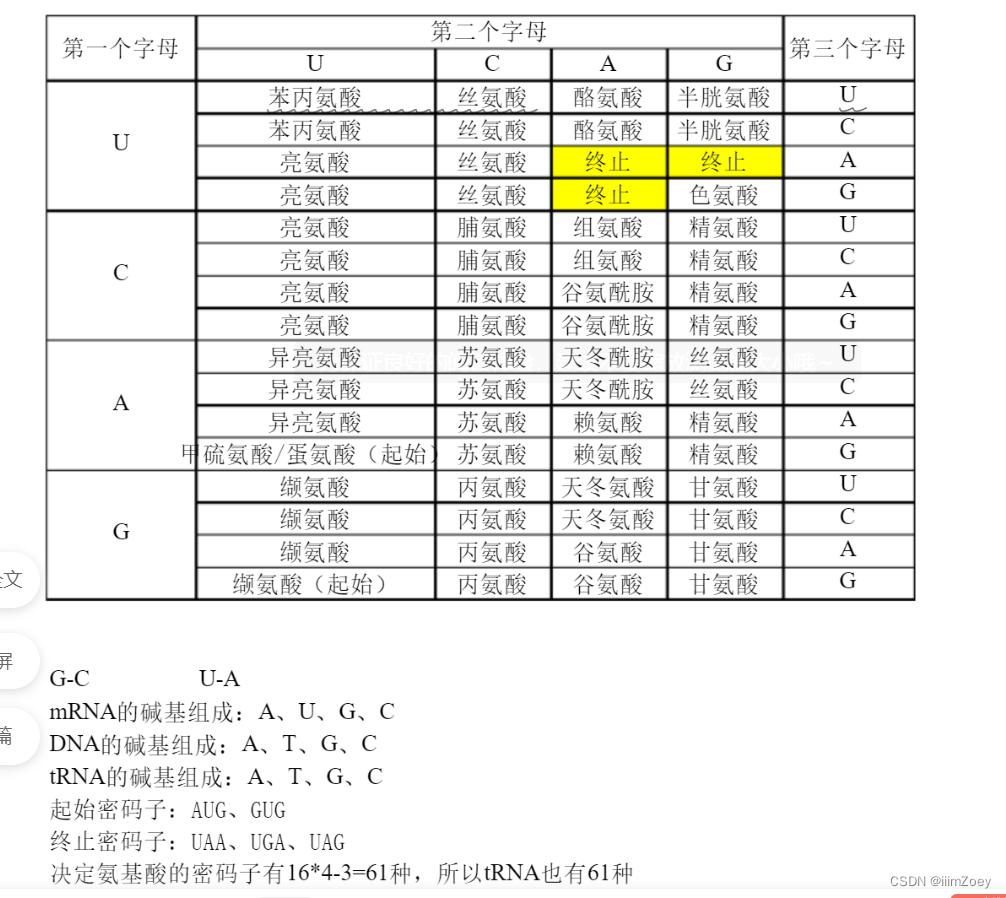
中心法则:DNA->RNA->蛋白质,中心法则说明,DNA可以转录形成RNA,RNA再翻译成一个个氨基酸,最后组合形成蛋白质
碱基对应氨基酸:
alphafold2论文精读(limu)
## 摘要
问题
蛋白质对于生命来说是必要的, 了解蛋白质的结构有助于理解蛋白质的功能
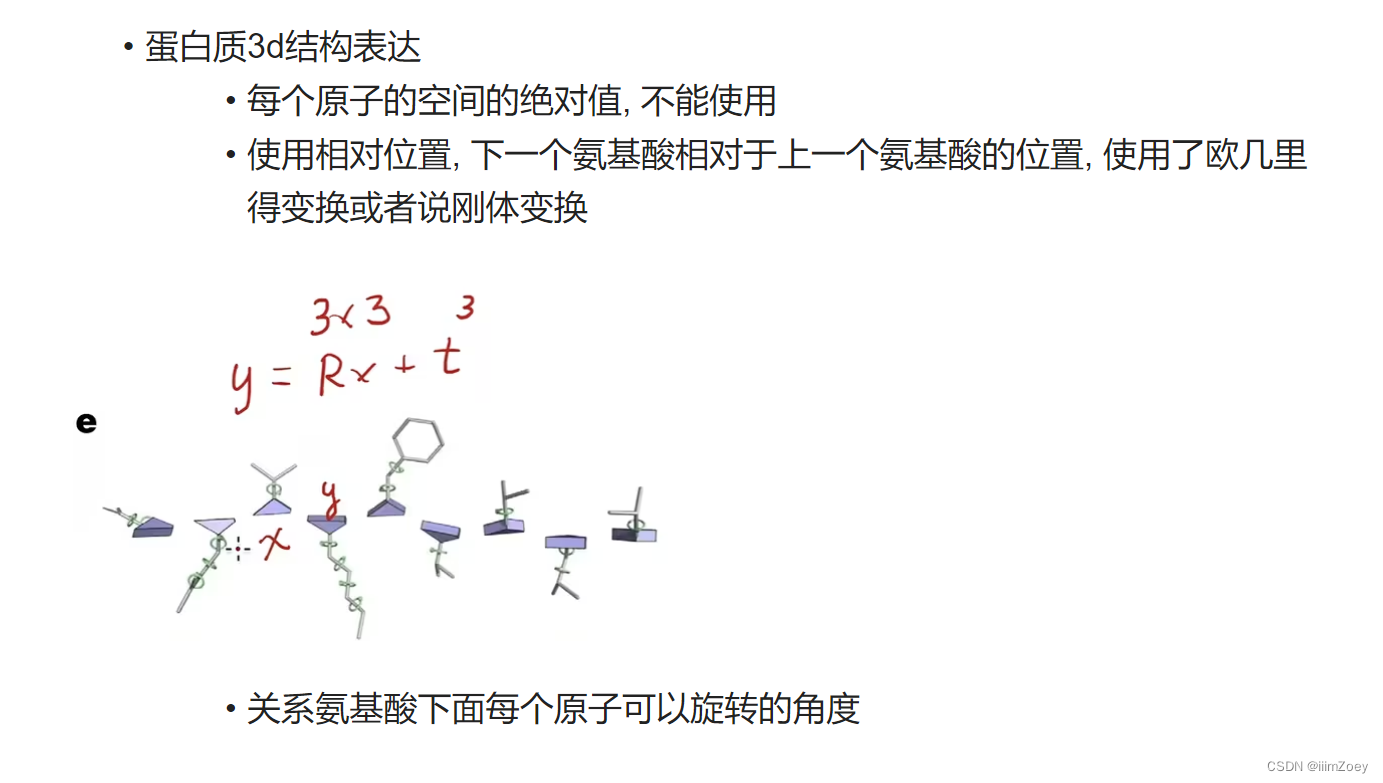
蛋白质是长的氨基酸序列, 不稳定, 容易卷在一起, 从而形成独特的3d结构, 从而决定了蛋白质的功能
预测的困难(蛋白质折叠问题), 只知道很少一部分蛋白质的结构, 通过显微镜不同角度观察蛋白质还原3d结构(冷冻方法cryo-em)观察费时费力
现有方法
AlphaFold1 精度不够, 不在原子的精度(氨基酸位置的预测 实验真实的位置偏差不在一个原子大小级别,这是非同源蛋白?同源蛋白预测比较简单有template)
AlphaFold2 能够达到原子的精度
AlphaFold2 使用了物理和生物学的知识, 也同样使用了深度学习
应用型的文章
问题对于领域来说重不重要
结果的好坏, 是不是解决了这个问题
找新问题或者开发新模型
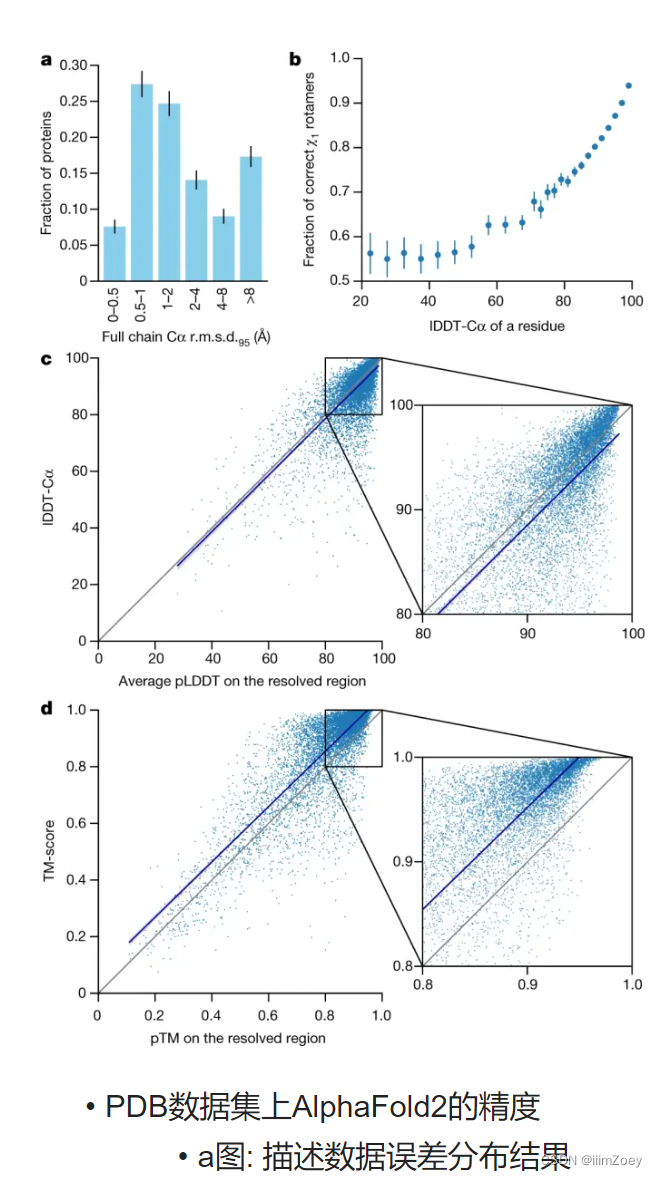
a图
x轴: 参赛队伍
y轴: 特定置信区间内, 预测的位置和真实的位置平均区别(单位是埃)
AlphaFold是1A, 碳原子的大小大概是1.5A 1A=10e-10
表述结果: 将绝对值变为相对值, 从1A到原子精度(就算之后有人达到0.5A,但是我先达到原子精度), 例如图片识别精度比别人提升10% 与图片识别比人类还要好,后者更有意思,因为大家只能记住里程碑的事情,
b,c,d图, 实验室和计算出来的结果, b错误率比较小, c和相对值进行比较, e复杂的图形
e图, 模型大概
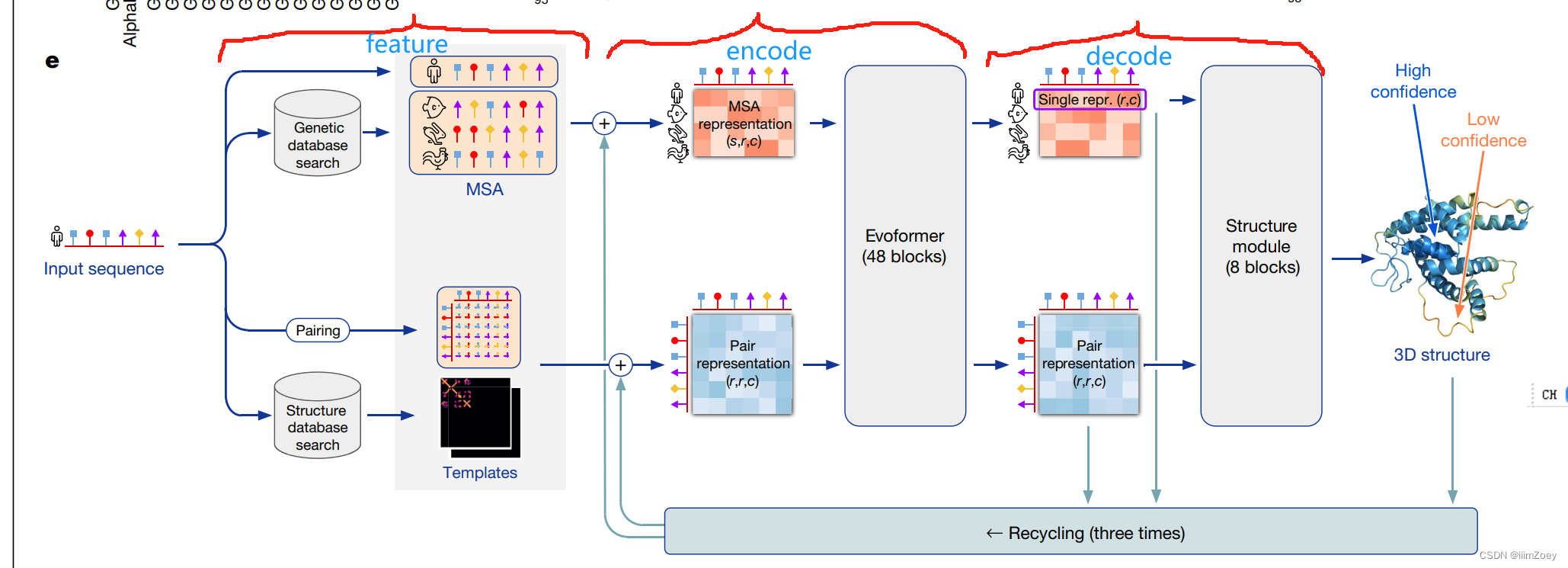
模型和训练
输入: 蛋白质氨基酸的序列
输出: 对每个氨基酸序列预测三维空间位置
可以大致分为三部分
第一部分: 抽特征, 主要是两大类特征, 不同序列的特征以及氨基酸之间的特征
直接导入神经网络
从基因数据库里搜索相似的序列(MSA, 多序列比对), 可以认为是字符串匹配过程
氨基酸直接的关系(Pairing), 其中是每个是一对氨基酸之间的关系, 最好是一对氨基酸在三维空间的距离(但是不知道)
结构数据库中搜索序列, 真实的氨基酸对之间的信息, 得到很多模板
第二部分: 编码器
将前面抽取的特征和后面的东西拼起来进入编码器
输入是两个三维的张量
MSA 表示(s,r,c)
Pair 表示(r,r,c)
s行(第1行是要预测的蛋白质, s-1为数据库匹配得到的)
r个氨基酸
长度为c的向量表示每个氨基酸
进入Transform的架构(Evoformer)
关系不再是一维的序列而是二维的矩阵
输入的是两个不同的张量
通过48个块抽取特征
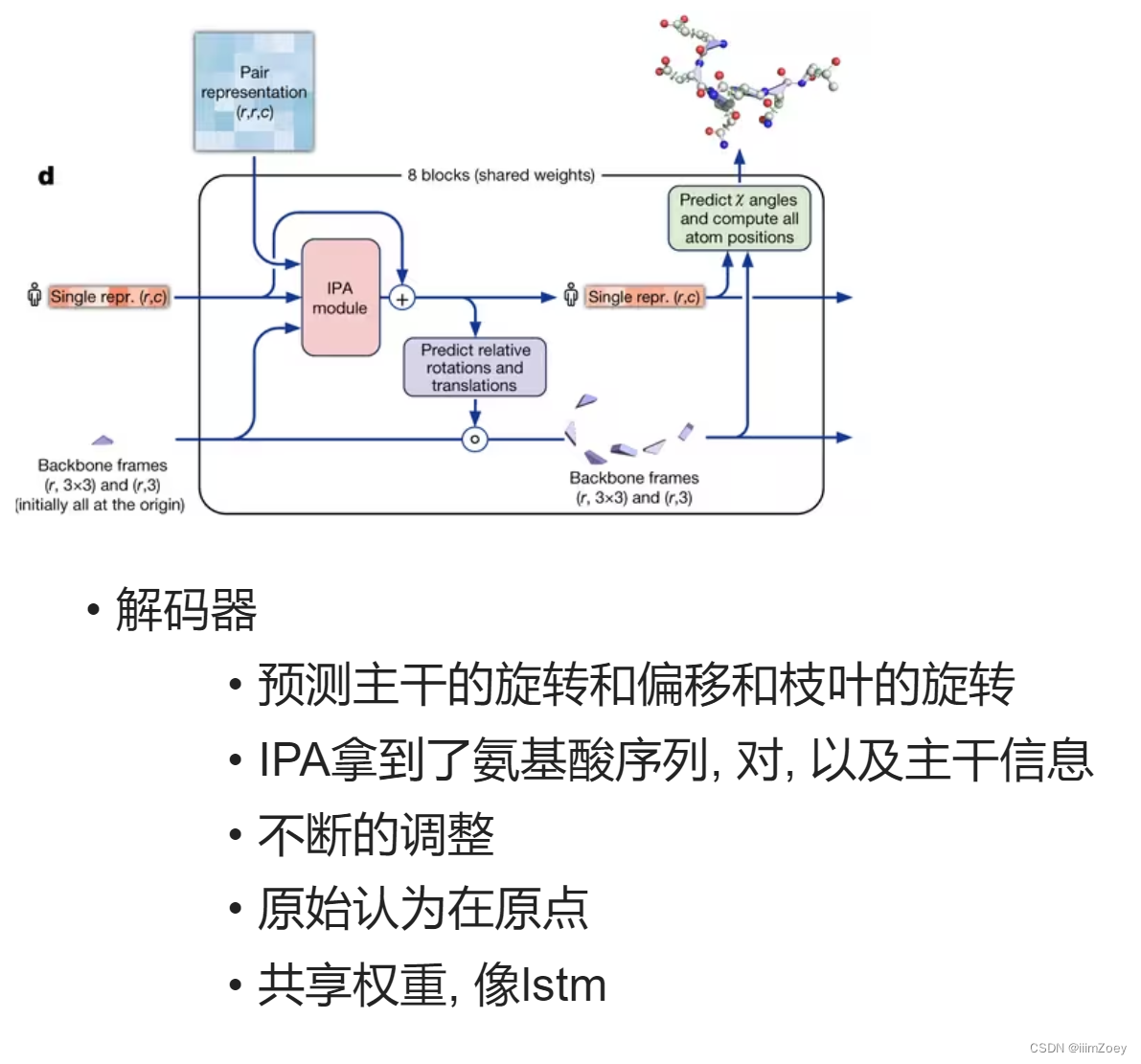
第三部分: 解码器
解码器拿到编码器的输出, 目标氨基酸的表示(r,c)和氨基酸对之间的表示(r,r,c)
回收机制
将编码器的输出和解码器的输出通过回收机制变成了编码器的输入, 迭代思想或者复制三倍深度更深
回收梯度不反传, 经过56层(48+8)就能计算梯度
encode:
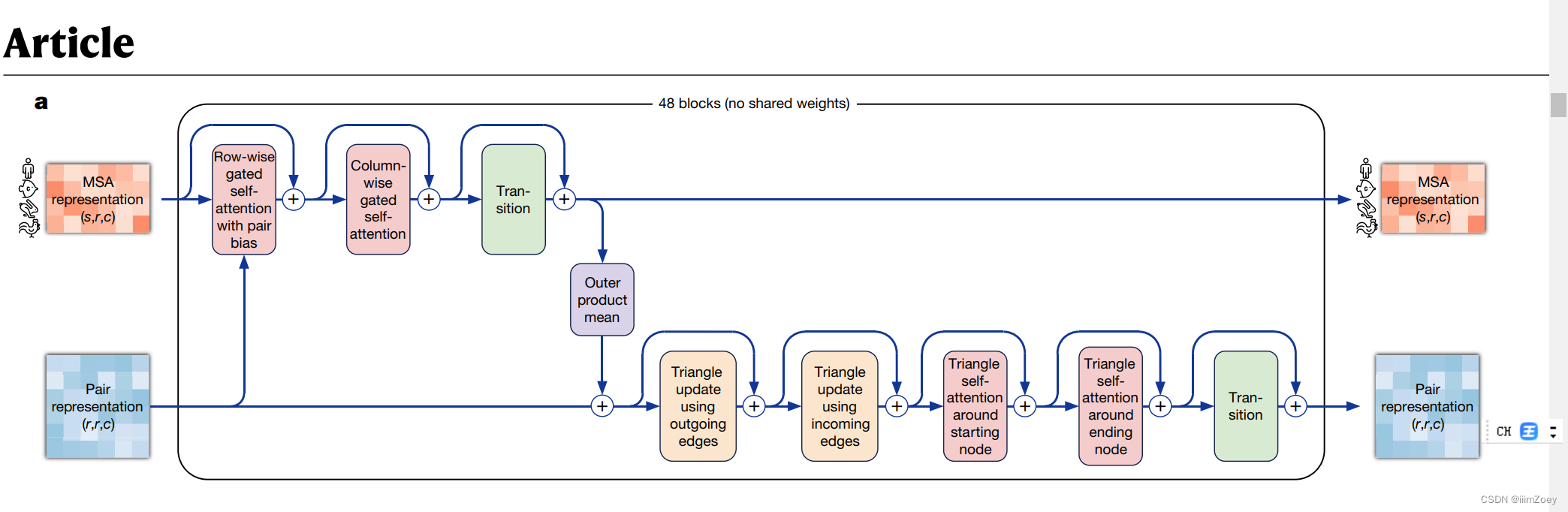

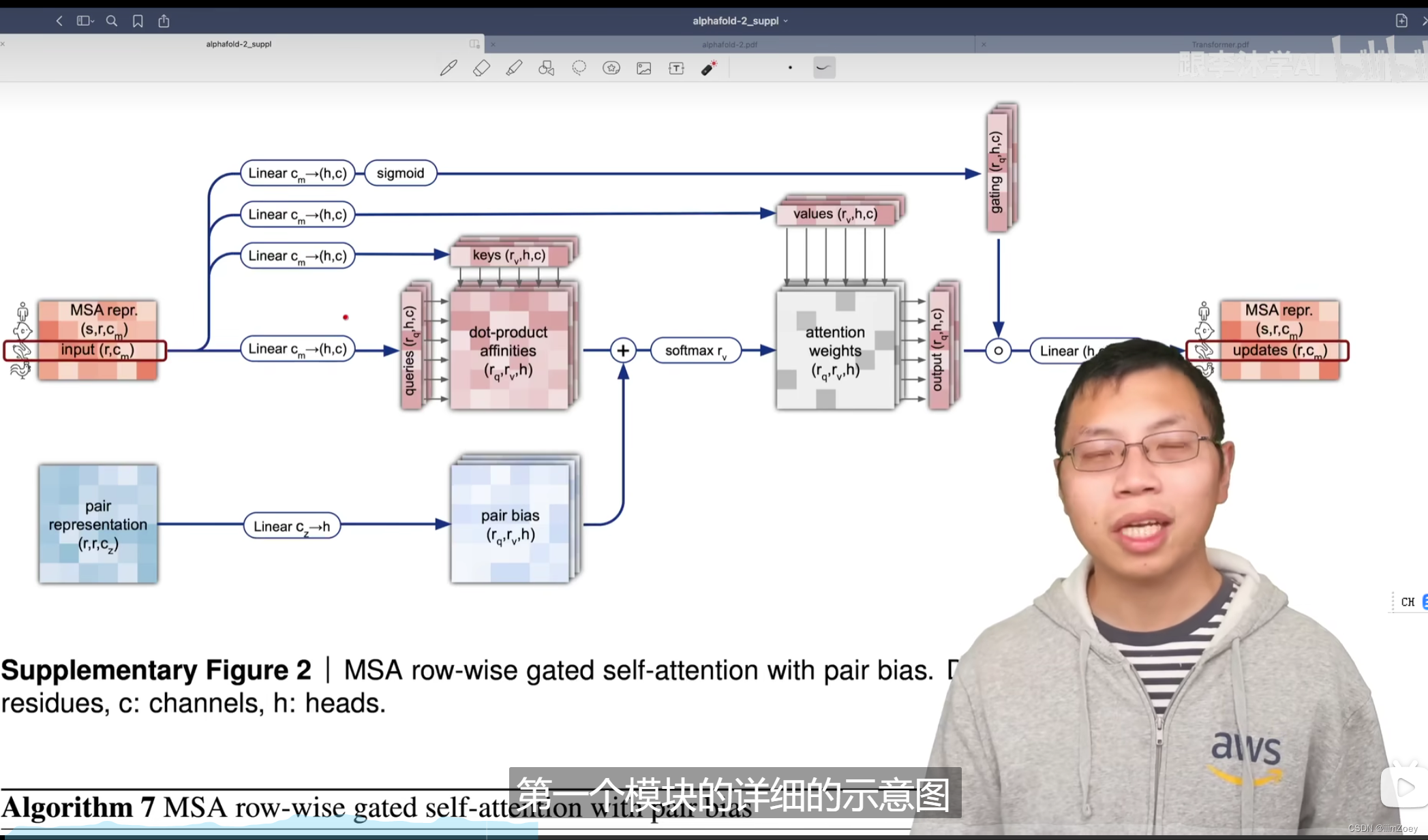

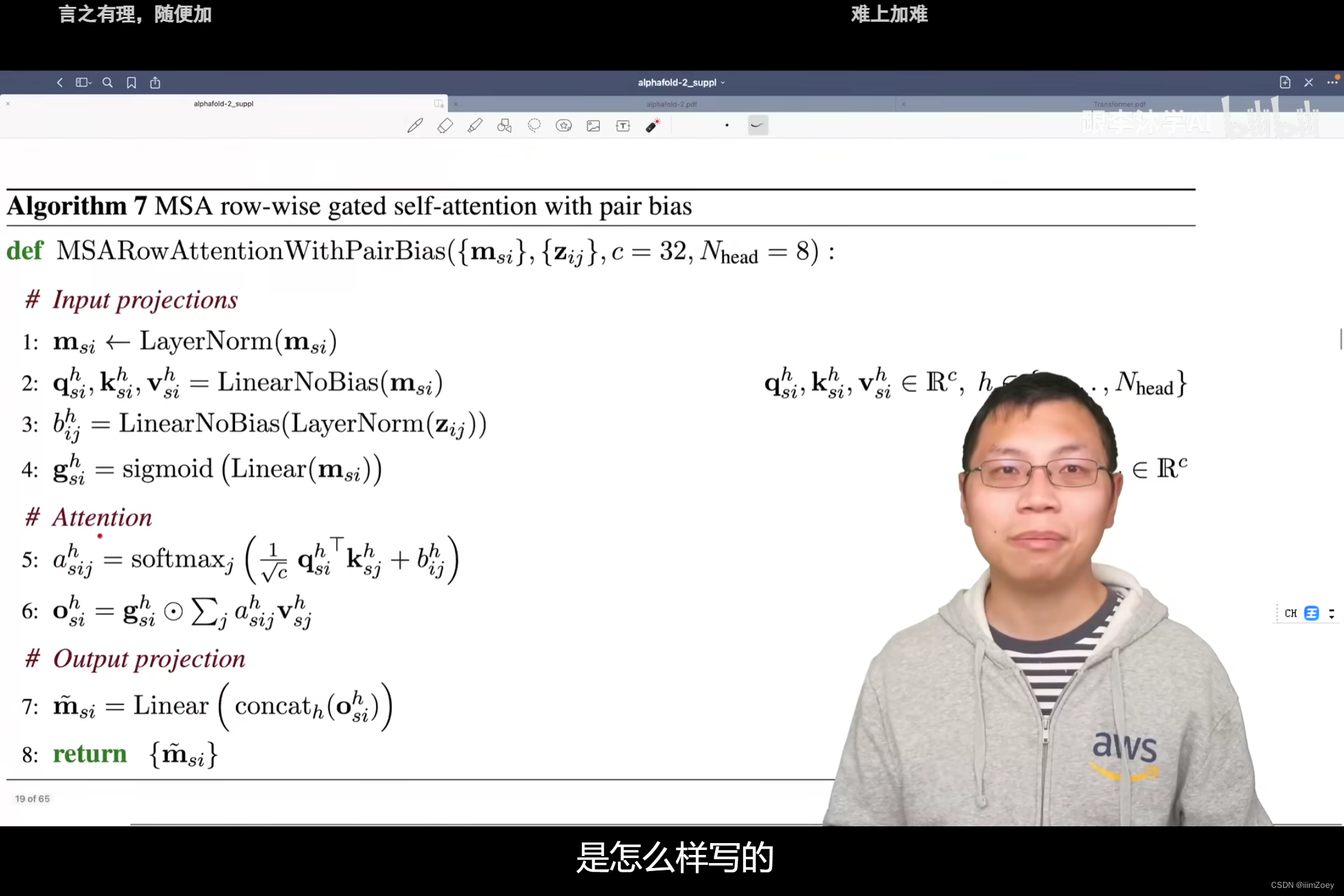
带门的:gated 如果是1则放出去;是0则不放出去
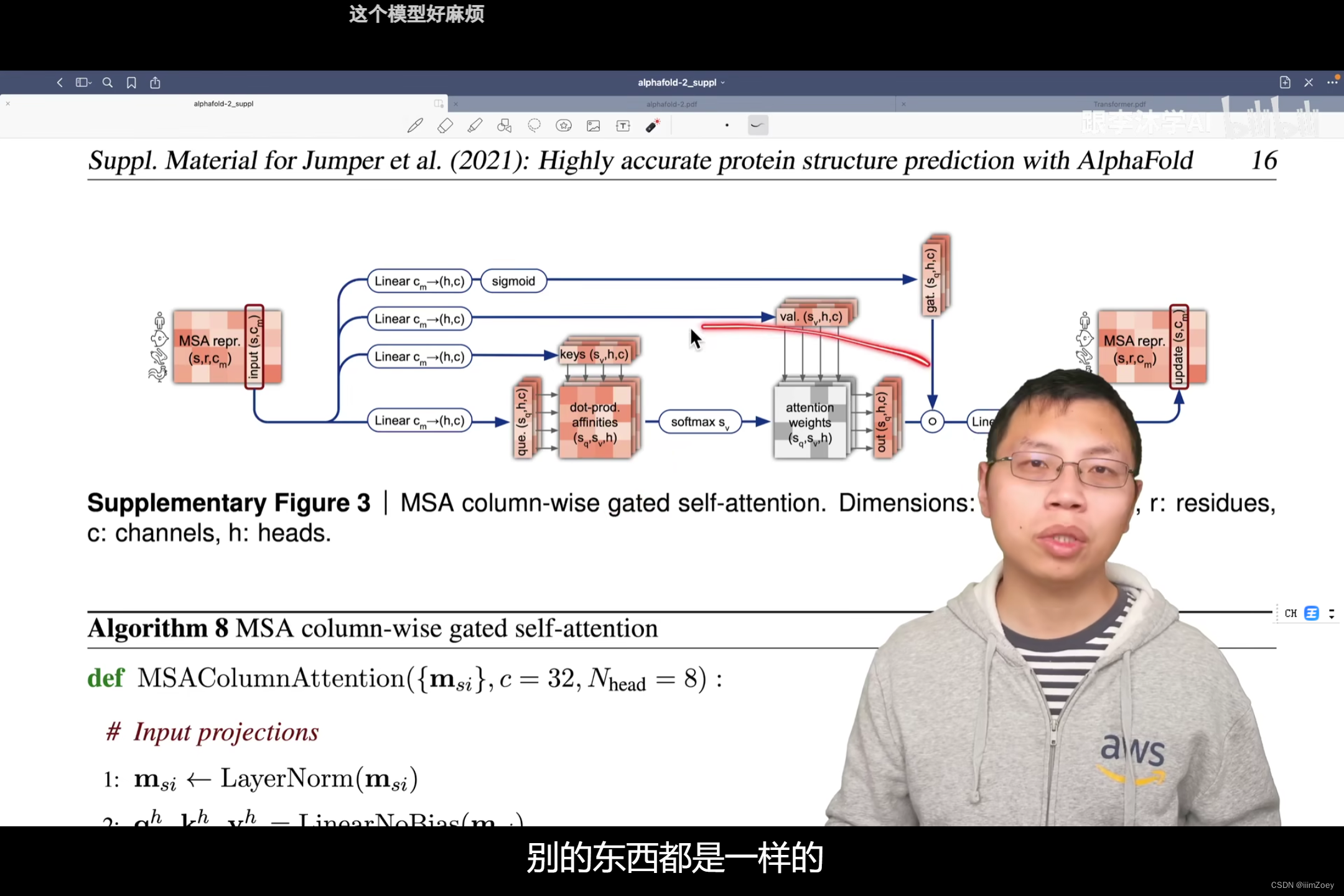
编码器每一块的第二部分:每列带门自注意力 编码器第二个模块(MSA column-wise gated self-attention)
和之前的区别: 按列, 无对信息做偏移加入
相当于transformer模型中 的MLP模块,将每个氨基酸矩阵拉成向量


以上是对编码器中MSA里面空间信息的建模
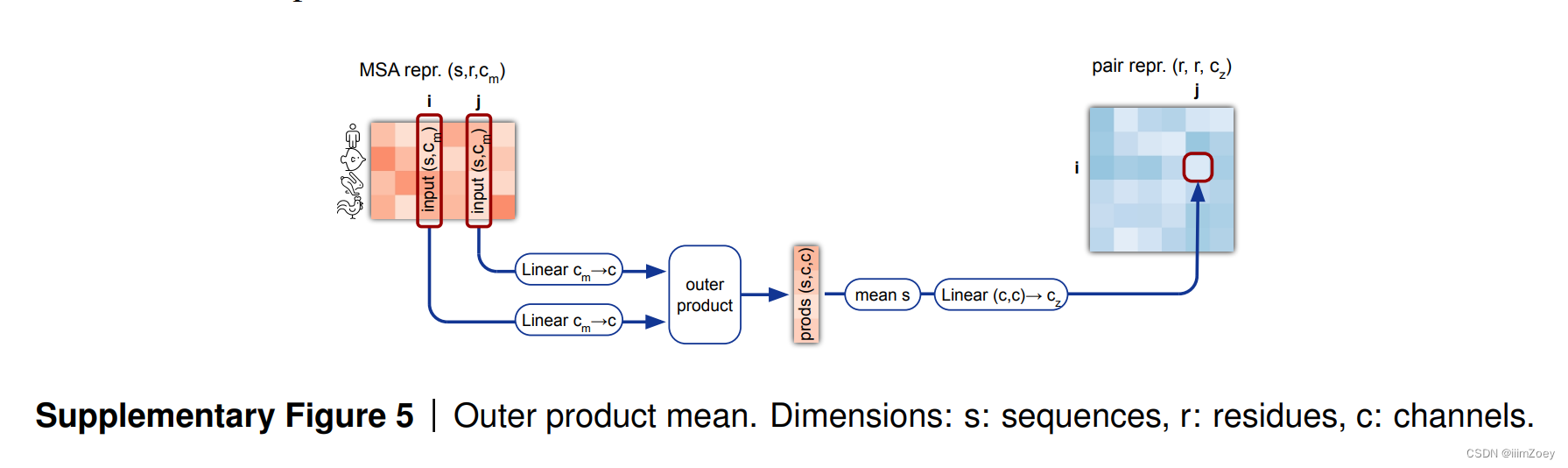
对pair的空间信息建模:


解码器
### 其他内容
其他的详细内容
特征如何抽取, 序列信息有长有短, 搜索到的也有长有短
如何将位置信息放入, transform出了名的对位置信息不敏感
回收机制如何执行
损失函数
主损失函数: FAPE, 根据预测的变换将蛋白质还原, 对应的原子在真实中和预测出来的位置, 这两距离的相减
使用没有标号的数据
使用noisy student self-distillation
核心思想: 先在有标号的数据集上训练一个模型, 然后预测未标号的数据集, 将其中置信的拿出来和有标号的组成新的数据, 重新再训练模型
核心关键点: 加噪音, 防止错误标号进入训练级. 加入噪音, 例如大量的数据增强, 甚至把标号改变, 模型能够处理这些不正常的标号
BERT
任务: 随机遮住一些氨基酸或者把一些氨基酸做变换, 然后像BERT一样去预测这些被遮住的氨基酸
训练时加入上述任务, 整个模型对整个序列的键模上更加好一点
训练参数
序列长度是256
batchsize是128
128个TPUv3上训练
初始训练7天, 额外的微调还需4天, 计算两属于中等
最大的问题是内存不够, 几百GB
预测性能(以V100为准)
256个氨基酸, 5min
384个氨基酸, 9min
2500个氨基酸, 18h
总结
编码器是在transformer上的改进,要处理两个不同输入, 每个输入在行和列上都有关系,对MSA representation和Pair representation两个模块不断进行数据的交互;
在编码之后进入解码器,解码器根据信息还原每个氨基酸在3D的位置,基于attention机制,显式的对位置信息进行了建模;(像RNN->编码器叫evoformer/ transformer)















 1156
1156

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









