







基于 MSA作为输入:
AlphaFold2
RoseTTAFold
ColabFold
ColabFold 通过将 MMseqs2 的快速同源搜索与 AlphaFold2 或 RoseTTAFold 相结合,加速预测蛋白质结构和复合物。ColabFold 的搜索速度提高了 40-60 倍,并优化了模型利用率,每天可以在具有一个图形处理单元的服务器上预测近 1,000 个结构。与 Google Colaboratory 相结合,ColabFold 成为一个免费且可访问的蛋白质折叠平台。
OpenFold
OpenFold 是 DeepMind 的 AlphaFold2 的 PyTorch 复现版本,用于自动处理蛋白质折叠实验。OpenFold 并不是同类中的第一个,但它是迄今为止最完整的,并且拥有与 AlphaFold 相等或更高的能力。与 AlphaFold 一样,以自己的名义,OpenFold 是完全开源的,并且在非常宽松的许可下提供。两者的参数都可以轻松下载并在 CC BY 4.0 下获得许可,而通过 GitHub 提供的代码在 Apache 2.0 下获得许可。这意味着任何有兴趣的人都可以将 OpenFold 用于几乎任何目的。
FastFold
蛋白质结构预测是结构生物学领域理解基因翻译和蛋白质功能的重要方法。AlphaFold 将 Transformer 模型引入了具有原子精度的蛋白质结构预测领域。然而,AlphaFold 模型的训练和推理由于其特殊的性能特点和巨大的内存消耗,既耗时又昂贵。在本文中,作者提出了 FastFold,这是一种用于训练和推理的蛋白质结构预测模型的高效实现。FastFold 包括一系列基于对 AlphaFold 性能的全面分析的 GPU 优化。同时,通过 Dynamic Axial Parallelism 和 Duality Async Operation,FastFold 实现了高模型并行缩放效率,超越现有流行的模型并行技术。实验结果表明,FastFold 将整体训练时间从 11 天减少到 67 小时,并实现了 7.5-9.5 倍的长序列推理加速。此外,我们将 FastFold 扩展到 512 个 GPU,并以 90.1% 的并行效率实现了总计 6.02 PetaFLOPs。
基于 pLM
自监督的预训练语言模型大多都是采用的BERT的结构,也就是采用堆叠Transformer的encoder layer,并采用随机mask掉残基来预测被mask残基类型的训练方式来学习残基之间的依赖关系。一个比较有意思的发现是,模型深层的attention score 形成的matrix可能编码了蛋白残基之间的接触关系,当我们对不同层attention map做一个简单的logisitic regression,便能粗略的预测出蛋白质序列中哪些残基是相互作用的。
使用来自蛋白质语言模型 (pLM) 的嵌入作为输入:
ESM-Fold(无监督接触预测)
大型语言模型超越简单的模式匹配来执行更高级别的推理并生成逼真的图像和文本。虽然在较小规模上研究了针对蛋白质序列训练的语言模型,但随着规模的扩大,人们对它们对生物学的了解知之甚少。在这项工作中,研究人员训练了多达 150 亿个参数的模型,这是迄今为止要评估的最大的蛋白质语言模型。研究人员发现,随着模型的缩放,它们学习的信息能够以单个原子的分辨率预测蛋白质的三维结构。研究人员提出了 ESMFold,用于直接从蛋白质的单个序列进行高精度的端到端原子级结构预测。ESMFold 与 AlphaFold2 和 RoseTTAFold 对于语言模型可以很好理解的低困惑度序列具有相似的准确性。ESMFold 推理比 AlphaFold2 快一个数量级,从而能够在实际时间尺度上探索宏基因组蛋白的结构空间。
ESMFold 和 AlphaFold2 的区别
使用语言模型表示来消除对显式同源序列(以 MSA 的形式)作为输入的需要
esmfold用transformer代替了Evoformer in AlphaFold2(alphafold2的encoder,input是MSA和氨基酸pair,evoformer因为处理两个input,是transformer的变形)
- 预训练的 Language Model 结构上和 Transformer Encoder 类似,但是num layers、hidden units有一定区别;
- 对Postion Embedding做了修改,可以支持更长的氨基酸序列编码(参考:苏剑林:Transformer升级之路:2、博采众长的旋转式位置编码);
- 基于ESM2的蛋白质结构预测模型ESMFold,取得了和AlphaFold2相当的效果,并且效率大幅提升(单卡V100预测一个384长的蛋白质氨基酸序列的结构,仅耗时14.2秒,传统MSA需要半小时)。
训练
数据集:我们在 PDB 链的不同子集上训练 ESMFold,并通过 AlphaFold2 预测的 12M 结构数据集进一步增强
esmfold用的
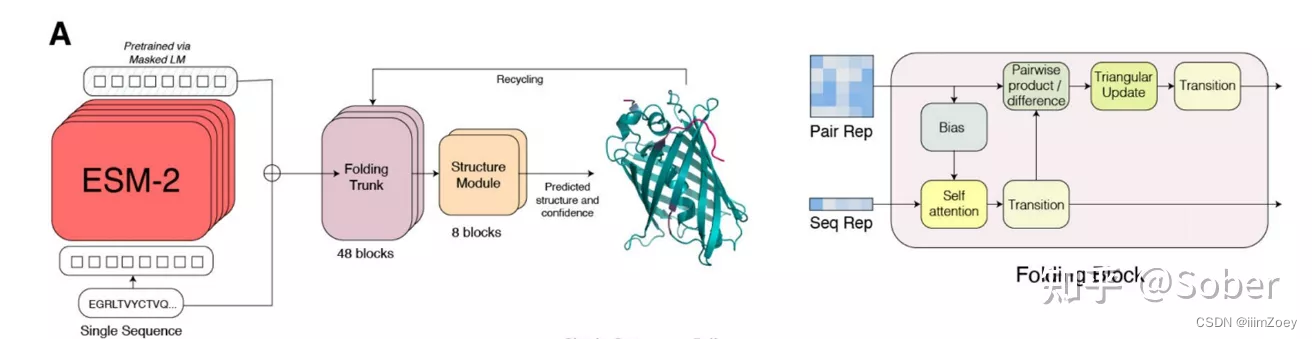
ESMFold的结构
作者将预训练好的语言模型ESM-2的蛋白质序列embedding和attention map接入与48层folding trunk和8层Structure Module来预测蛋白质全原子的结构。这里structure module与AF2相同,而folding trunk是退化版的evoformer,因为只有单序列所以axis attention 机制就退化成了普通的self-attention,而节点与边embbeding的更新方式保持相同。 值得注意的是,ESMFold训练时不仅使用了PDB数据库中实验解析的单链结构,还使用了AF2预测的高置信度的蛋白结构。
pytorch实现
1.hugging face调用pretrained模型(colab上用)
https://huggingface.co/docs/transformers/model_doc/esm
from transformers import AutoTokenizer, EsmForProteinFolding
model = EsmForProteinFolding.from_pretrained("facebook/esmfold_v1")
tokenizer = AutoTokenizer.from_pretrained("facebook/esmfold_v1")
inputs = tokenizer(["MLKNVQVQLV"], return_tensors="pt", add_special_tokens=False) # A tiny random peptide
outputs = model(**inputs)
folded_positions = outputs.positions2. colabfold has integrated ESMFold so that you can easily run it directly in the browser on a Google Colab instance.
https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/ESMFold.ipynb
3.ESM Metagenomic Atlas by Meta AI
接口
4.下载esmfold模型 源代码
4.1利用pytorch hub

import torch
model, alphabet = torch.hub.load("facebookresearch/esm:main", "esm2_t33_650M_UR50D")
# 这一行代码使用 PyTorch Hub 从 Facebook Research 的 esm 仓库中加载 ESM 模型。
# esm2_t33_650M_UR50D 是一个具体的模型标识符,代表 ESM 模型的一个变种。它是一个拥有 650M 参数模 #型
model = model.eval().cuda() #如果没有gpu会转为cpu 明确使用cpu:model.eval.cpu()
# Optionally, uncomment to set a chunk size for axial attention. This can help reduce memory.
# Lower sizes will have lower memory requirements at the cost of increased speed.
# model.set_chunk_size(128)
sequence = "MKTVRQERLKSIVRILERSKEPVSGAQLAEELSVSRQVIVQDIAYLRSLGYNIVATPRGYVLAGG"
# Multimer prediction can be done with chains separated by ':'
with torch.no_grad():
output = model.infer_pdb(sequence)
with open("result.pdb", "w") as f: #将推断结果保存到名为 "result.pdb" 的 PDB 文件中
f.write(output)必须有openfold,下载openfold和pip install deepspeed 都必须有cuda,cuda必须有gpu
CUDA是NVIDIA独有的GPU并行计算平台,必须有NVIDIAGPU才能运行CUDA程序。
colab的缺点
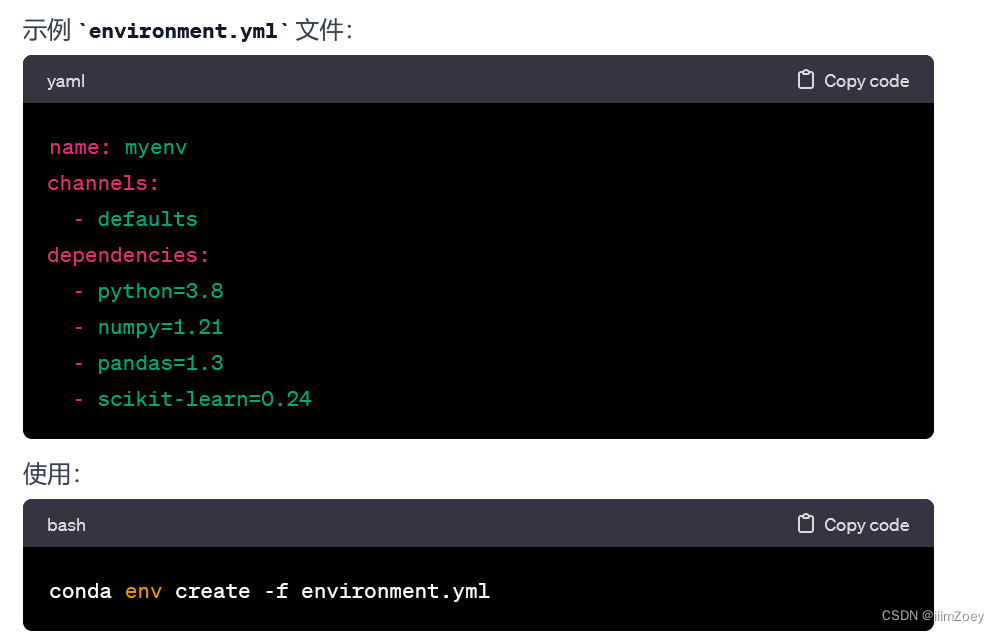
conda env create -f environment.yml.和pip install -r requirements.txt的区别
conda env create -f environment.yml:
- 使用
conda包管理工具,适用于 Anaconda 或 Miniconda 环境。 - 从指定的 YAML 文件 (
environment.yml) 中创建一个新的 conda 环境。 environment.yml文件通常包含所有需要的软件包和其版本,以及其他环境配置信息,如 Python 版本和操作系统平台。- 能够处理 Python 包以外的系统级依赖项
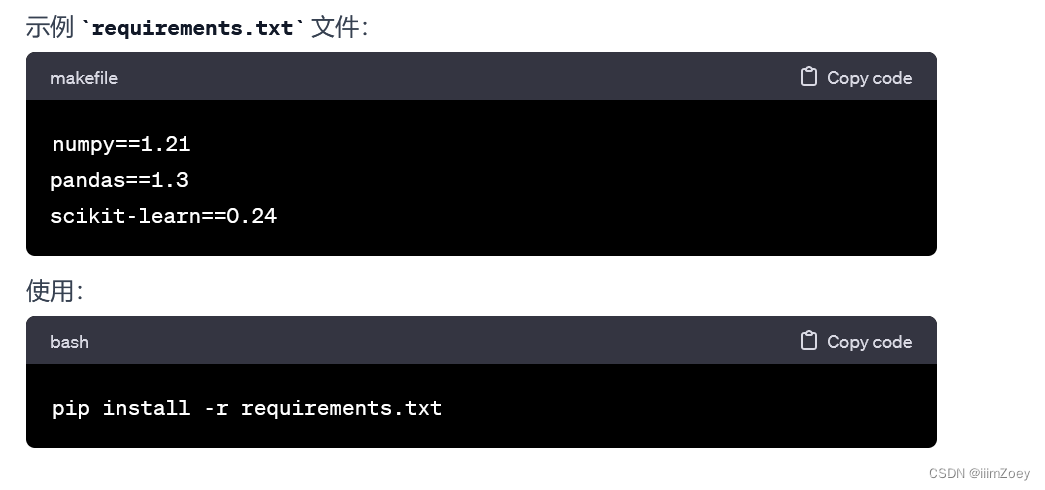
-
pip install -r requirements.txt:- 使用
pip包管理工具,是 Python 的默认包管理工具。 - 从指定的文本文件 (
requirements.txt) 中安装所有列出的 Python 包。 requirements.txt文件包含软件包名称及其版本,通常是通过pip freeze命令生成的。- 主要处理 Python 包,对于系统级依赖项可能需要其他工具。
- 使用
总体而言,conda 和 pip 都是流行的包管理工具,但它们在环境管理和软件包解析方面存在一些区别。conda 还能够处理非 Python 软件包,而 pip 更专注于 Python 软件包。选择使用哪个工具通常取决于您的环境和项目需求

















 6417
6417

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









