






Gaussian 09W和GaussView 5.0学习记录
Calculation setup
Job type
Energy:对已经给定的分子进行能量与性质计算
Optimization:对分子进行几何优化,在势能面上试图找到最低点
Frequency:计算基态或者激发态的振动光谱,鉴别势能面上驻点的性质,以及热力学性质,要在已经Optimization操作后才能计算频率
Opt+Fre:上述合二为一
Method
Method:
参数1:ground state基态
参数2(计算方法):semi-emp 半经验;har-fock自洽场方法;DFT 密度泛函;计算时间从上至下通常越来越长精度越来越高。
参数3(波函数类型的选择):restrict限制性闭壳层波函数;unre非限制性开壳层波函数;res-open限制性开壳层波函数。如果不设定就默认。
参数4(计算理论的选择)
Basis set(基组):一般来说,基组越往上,那它所适应计算的元素越靠近元素周期表的前端;涉及过度元素之类了,选择基组也应该往后走。后面的参数是考虑离散函数和轨域需要设定。
Charge:价数
Spin:自旋多重度。 它等于2S+1 ,S等于孤对电子

Title
给工作命名的,不重要
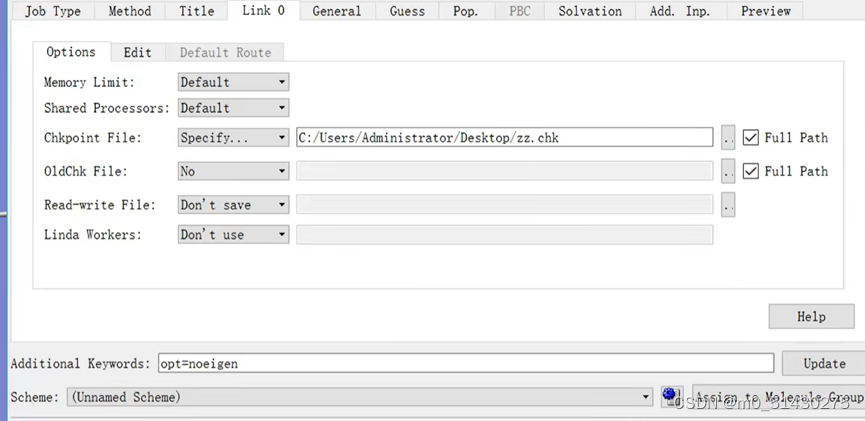
Link0
Memory list:内存限制,注意设大些
Chk-file:计算是耗时的,避免计算文件因为意外丢失。相当于断点。需要手动设置文件保存路径并命名
General and Guess:对分子结构的一些猜测。一般不用管
Share processors:分配的电脑核心数
NBO:NBO相关计算
PBC:做晶体的有关计算
Solvation:溶剂化效应的选择。Modle对模型选择;solvent:溶剂的选择
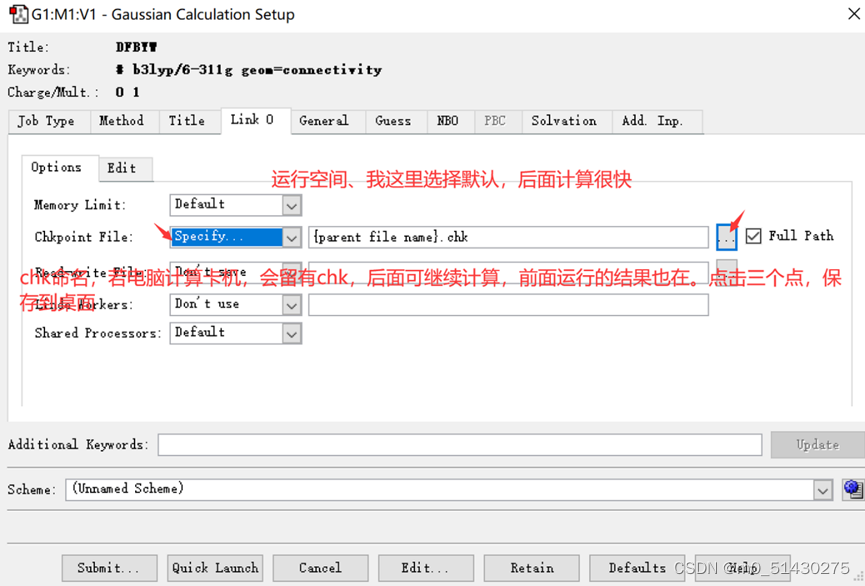
Additional keyword:用于添加便于区分文件的关键字
Schme:添加设置的保存。避免重复调整各种参数
最底下一栏
Retain:保存当前参数设置。
Edit:通过外部的编辑器对输入内容进行修改
Submit:直接提交
Quicklaunch:点击后会直接开始在gaussian进行计算
Guassian 09
File
创建一个新的输入文件or打开一个创建好的输入文件
Process
理解为用于控制当下的执行任务
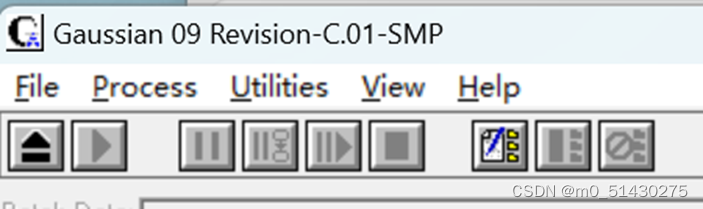
左1:打开已有或者已保存的文件
左2:开始运行当前的文件
中1:暂停当前任务
中2:
中3:恢复执行当前的任务
中4:终止当前任务
右1:批处理文件相关。编辑或者建立批处理。
右2:在当前作业完成后终止批处理
右3:终止当前的作业和批处理

1:打开外部编辑器(wps、notepad)
2:编辑输出文件
Existing File job Edit
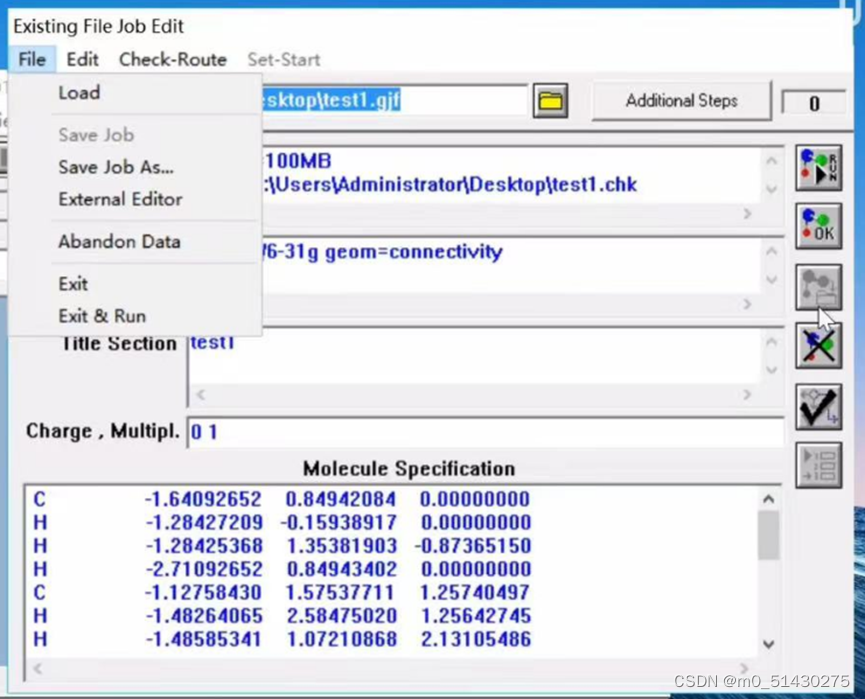
右1:退回到主目录并且开始执行任务
右2:退回到主目录但不会执行任务
%section:看到之前在gaussian view中设置的参数
Route Section:
Charge,Multipl:电荷和多重度的说明
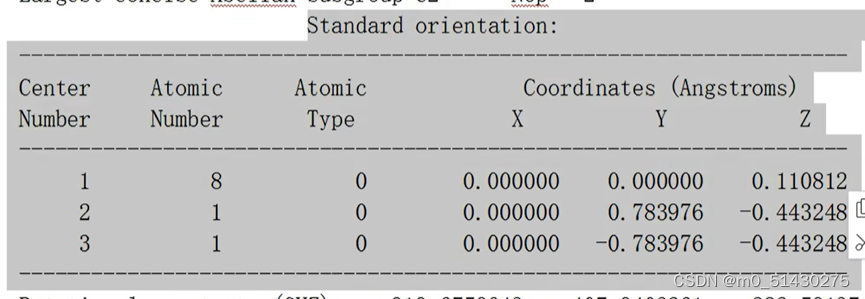
.out文件
标准坐标,可以通过这个来转化集合构型
能量:

分子轨道能量:

电荷分布:

偶极矩:

R(1,2)表示序号为1,2原子之间的键长;A1(2,1,3)是指序号为2,1,3三个原子之间构成的键角。D(3,1,2,5)是3,1,2,5构成的二面角

下面这个部分是指在进行最小值寻找(优化计算),可以看到一共进行了25次:

列出了键长旧值和新值的比较,键长、键角、二面角:
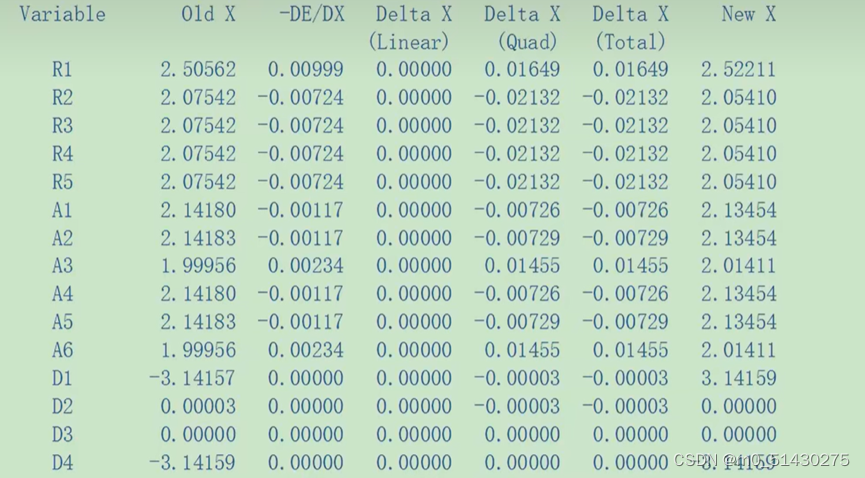
Converged:是否收敛,代表是否优化完成,即是否达到能量最低点

如下图所示:结果已经收敛。同时显示了预测结果的改变值:

通过几何优化,我们可以得到很多能量,我们需要的是经过最后一次迭代完成,前一次的能量
几何优化
算频率需要先对分子进行几何优化,opt+Freq选项试图找到势能最低点

在输出文件中可以看到震动频率和震动强度的信息:
1.震动频率 2.优化质量 3.力场数 4.IR强度
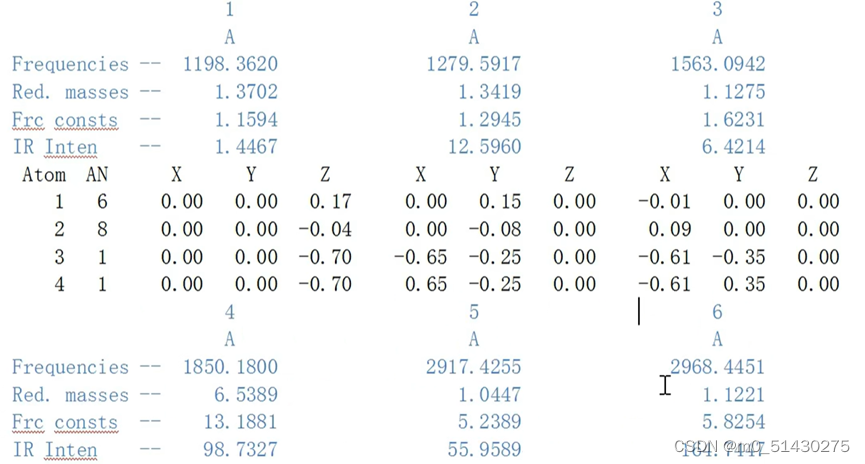
下图所示的信息:
- 电子总能量与零点能之和
- 内能(热力学能)
- 焓
- 自由能
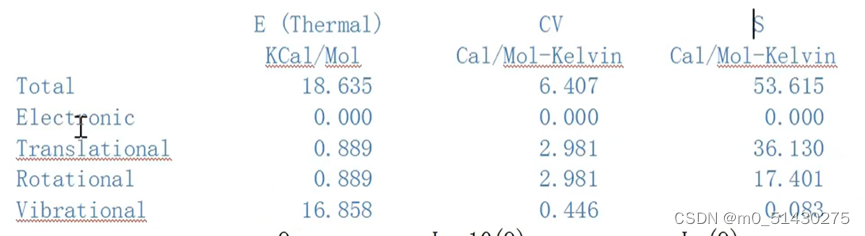
 热化学数据:
热化学数据:
分别是:电子能 平动能 转动能 振动能
在gaussian view里面可以打开.out文件,然后点display vibration可以看到预测的光谱图

优化计算
先在chemdraw里面画好图,然后在chem3D里面优化分子立体构型。对于大分子,最好使用半经验模型预优化,然后再采用DFT进行计算。
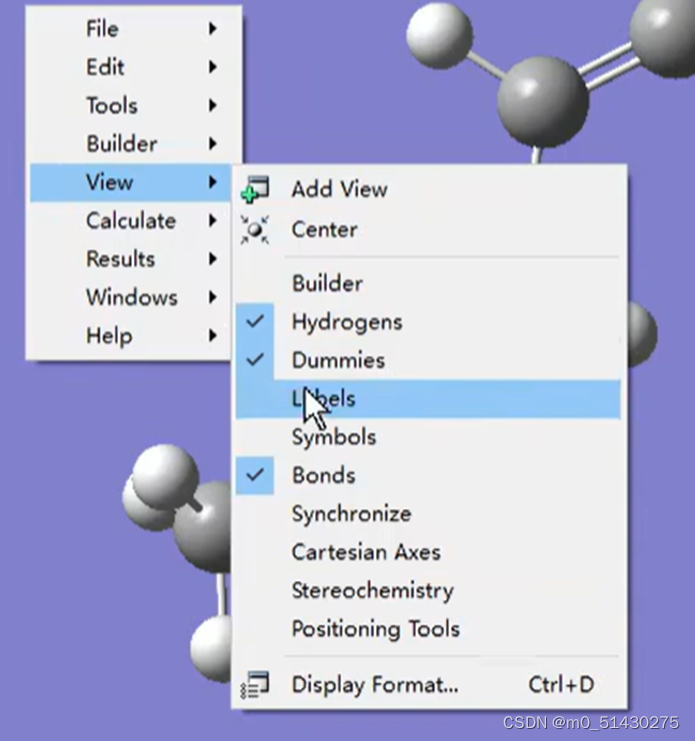
在输出结果内,view-labels可以看到原子序号数:
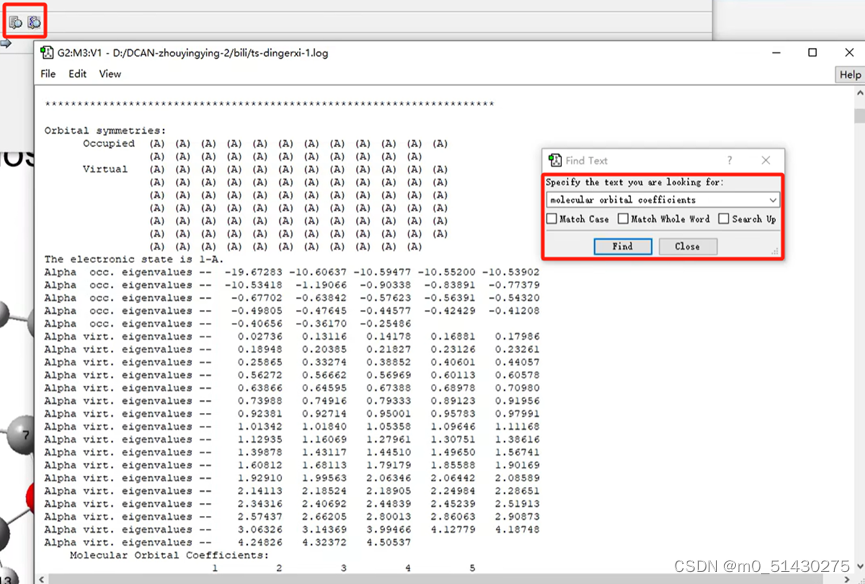
如图所示,点击左上角的放大镜logo可以看到输出文件,然后ctrl+f搜索:molecular,查看HOMO/LUMO轨道能级图。
一个分子的HOMO轨道和另一个分子的LUMO轨道之间能级差越小越容易反应
如下图所示:两个分子HOMO/LUMO轨道能级差更小的一组来决定反应。并由HOMO方提供电子,LUMO方提供空轨道接收电子。
能隙=LUMO-HOMO


对电子密度进行研究、绘图
打开chk文件,然后右键Results-surface:
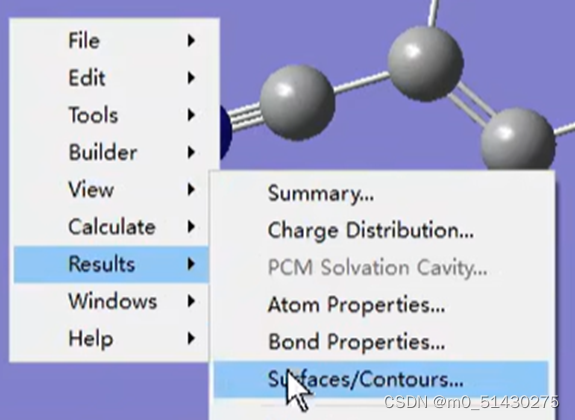
Cube actions选择total density:
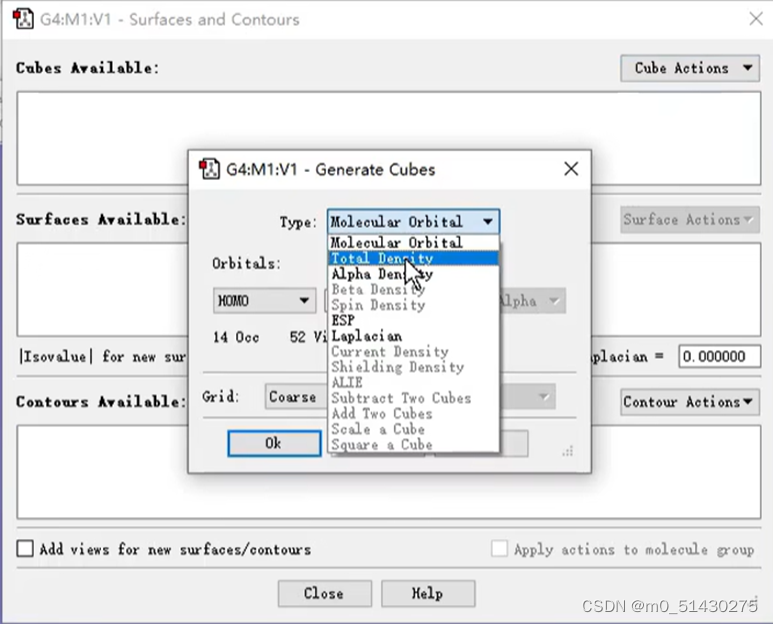
再将它的密度改为0.002:

再surface actions中选择new mapped surface:

直接点击OK就行:
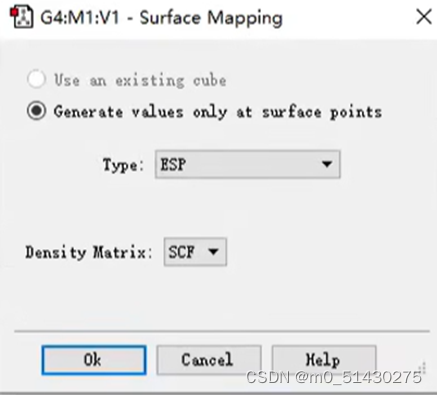
设置完后,点击view-display format,设置:
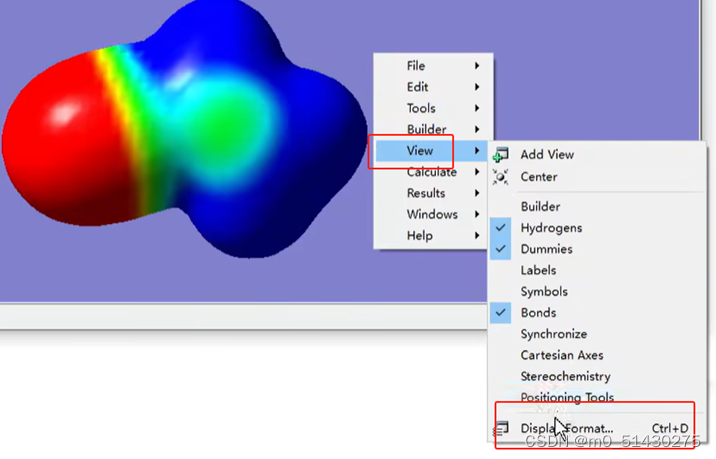
在界面中选择surface,format中修改为transparent(透明),下面拉条可调整不透明度:
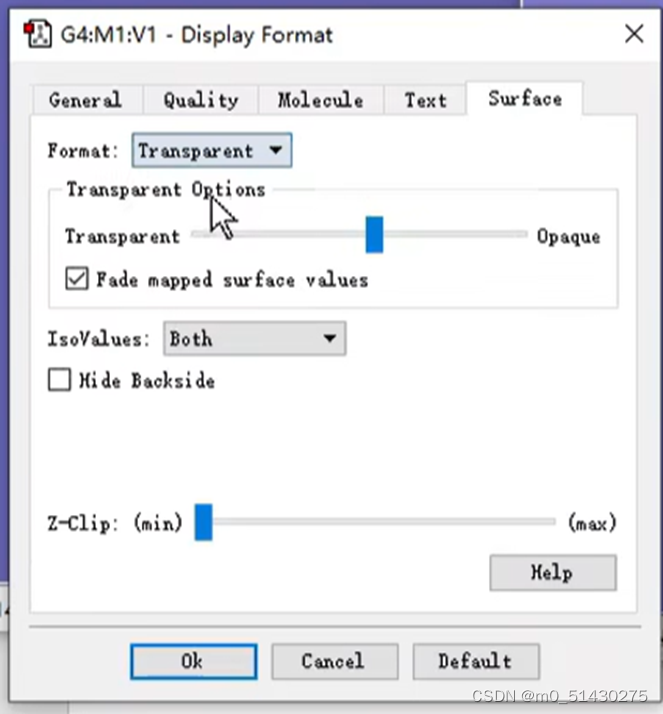
得到如下图所示的结果:
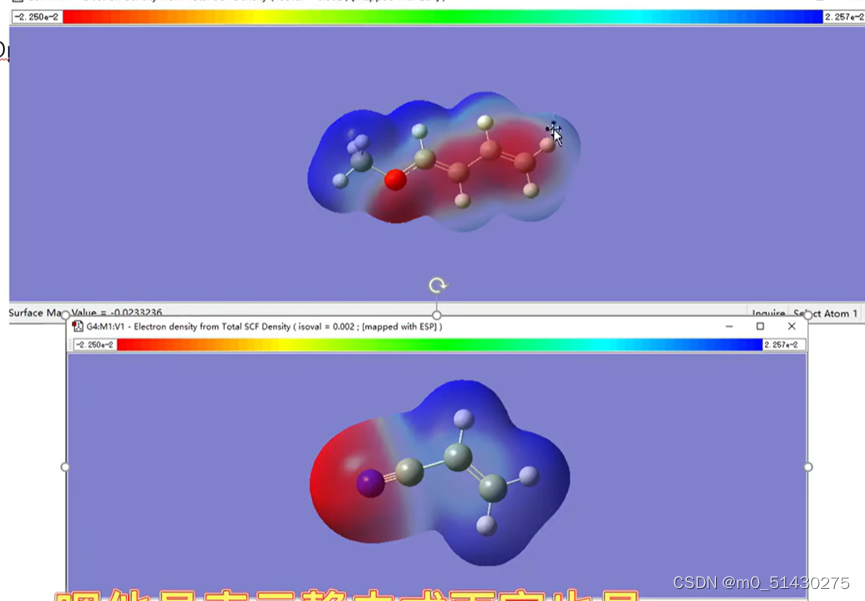
红色区域是代表偏负电荷区域,蓝色代表偏正电荷区域。根据之前判断出来的:HOMO方提供电子,LUMO方提供空轨道接收电子。
根据电子云的浓度(即颜色的深度)来初步判断可能的反应位点
计算分子的HOMO/LUMO,判断反应位点
先用opt+fre跑一遍:参数选择DFT,save生成gjf文件,然后在gaussian里面跑。接着把跑出来的.out文件拖到gauss view里设置,改为算Energy。用DFT方法需要加em=gd3bj 优化方法。

查看HOMO/LUMO轨道能级,并将分子轨道图设置为白色背景:
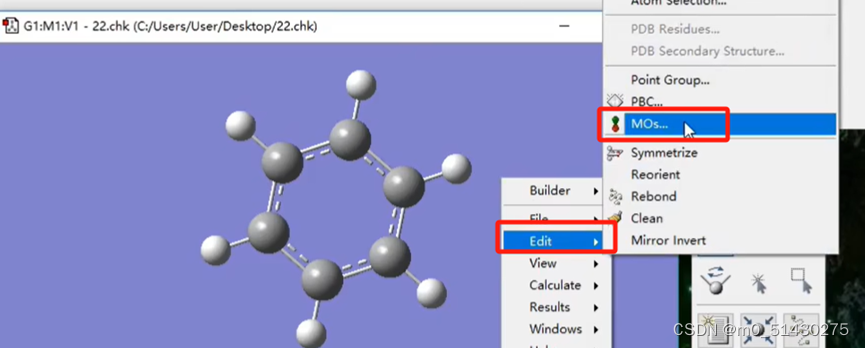
在chem darw里面画的图需要保存为gjf或者MDL MolFile 的mol形式

将输出文件往下拉,ctrl+f搜索molecular orbital coefficients还能看到每个轨道的贡献:

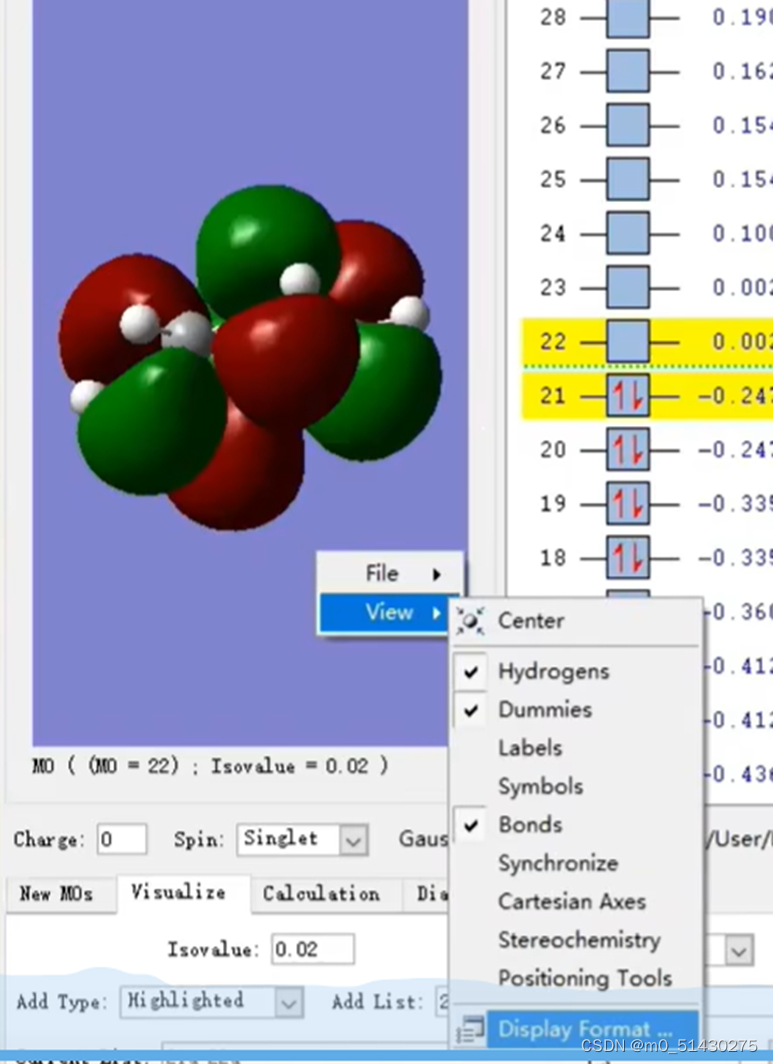
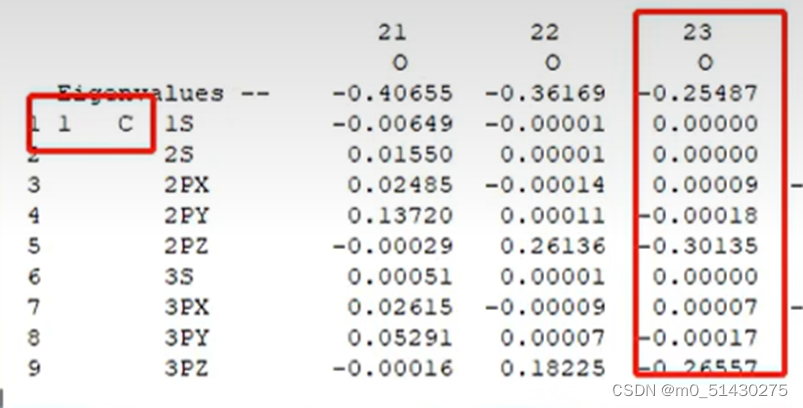
承接上节:在研究了反应位点后,比如知道了HOMO是第23条轨道,就去找第23条轨道(横坐标)。左上角是碳原子编号数。纵坐标是某某号碳原子的轨道贡献:
同理,还需要研究另一个分子的LUMO轨道,打开如下图所示界面:
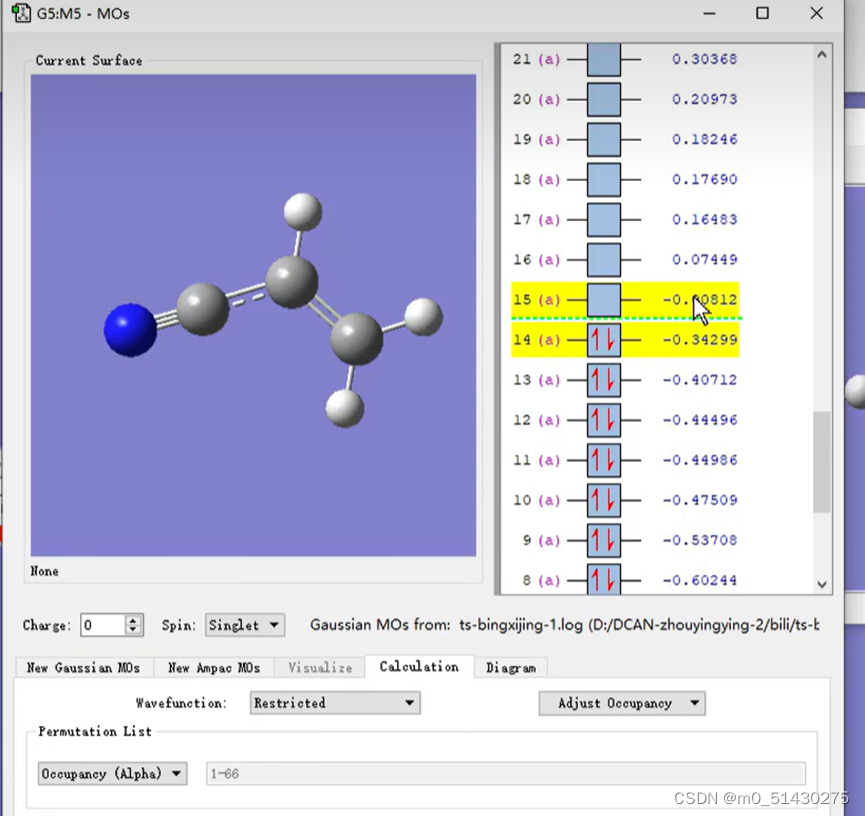
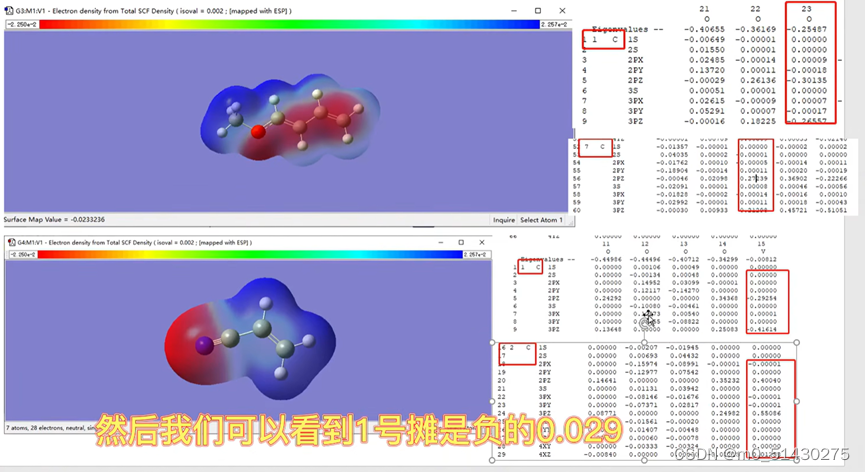
可以看到,提供LUMO轨道反应物的LUMO轨道序号为15,因此待会去.log文件里面需要找第15号轨道。然后根据之前分析出的电子云的浓度(即颜色的深度)来初步判断可能的反应位点的结果,来分析15号碳原子的某两个反应位点碳原子,比较它们谁的贡献更大,谁就是真正的反应位点。
预测过渡态的方法与理论

• 使用TS,你只需要提供想要的过渡态的初猜。
• 使用QST2,你需要提供反应物和产物的结构,并确保各个原子的序号是对应的。Gaussian将会自行搜索过渡态。
• 使用QST3,你需要提供反应物、产物的结构和过渡态的初猜。
上述提供反应物或产物的结构时并不需要事先优化。
过渡态(Transition state,TS) 是一个化学反应的反应坐标-势能图象中的极大值点。这么说听起来可能比较抽象,举个例子:
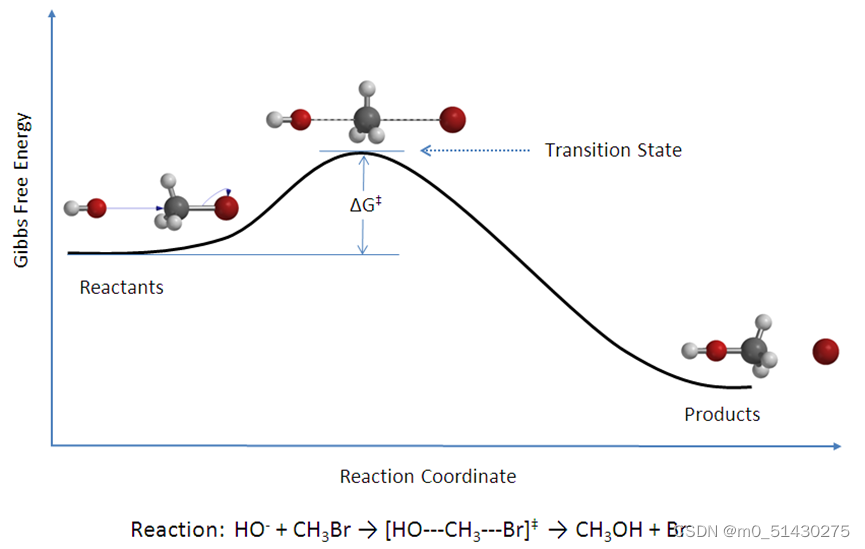
在一个化学反应的过程中,反应物走向产物的过程总会经历能量先升高后降低的过程,使得反应进行需要一定量的能垒(活化能),能量最高的状态就是过渡态。
如果将反应物和产物看成一个大体系的两种原子排布方式,两者都是势能面上的点,那么可以认为,这样的一个反应其实是从体系势能面上的一个极小值点翻山越岭到达另一个极小值点的过程:
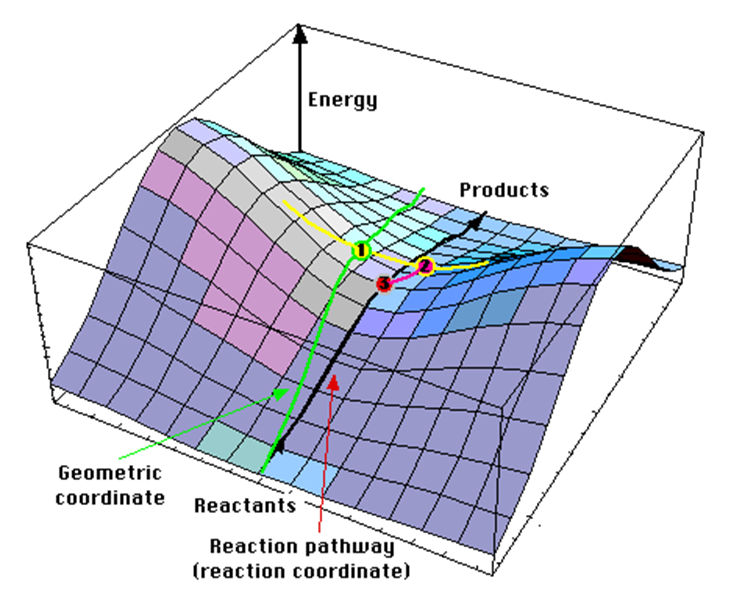
在这样的“地形”模式下,虽然两点之间存在无数条路径,但显然,反应中会采取爬坡能垒最小的路径。例如上图的黑线能垒就要比绿线能垒低;如果让路径沿着黄线滑动,在黑线时的能垒是最低的,是我们要找的最小能量路径(Minimum energy path,MEP),其最高点就是我们要找的过渡态。
因此,势能面上的过渡态具有以下特征:
在一个方向上是势能极大值点(黑线的最高点)
在其它方向上是势能极小值点(黄线的最低点)
数学上称这样的点为一阶鞍点,它属于上文曾提到过的驻点。这样,计算过渡态实际上就是在求算势能面的一阶鞍点,属于几何优化的一种。
QST2方法预测过渡态
使用QST2,你需要提供反应物和产物的结构,并确保各个原子的序号是对应的。Gaussian将会自行搜索过渡态。
以如下反应为例,介绍QST2方法:
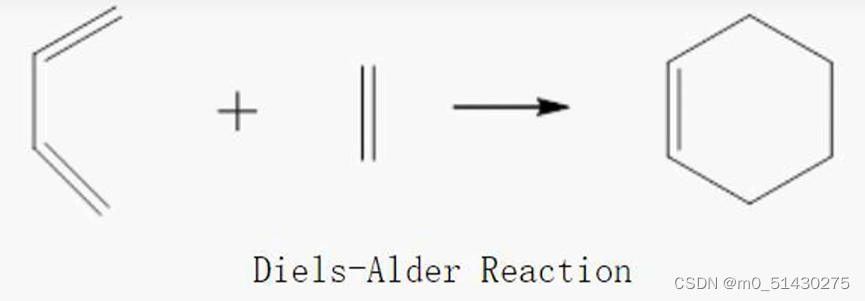 黄色原子按钮可以显示原子序号:
黄色原子按钮可以显示原子序号:
在gauss view中,先画出两个反应物,乙烯的11,12号碳原子进攻丁二烯的1,7号碳原子。需要先摆放好方向,有利于寻找过渡态:

接着又需要单独创建一个guass view窗口来画产物,在算过渡态的时候,要保证反应物与产物的原子编号保持一致。可以直接拷贝一份反应物,新建一个窗口用来画生成物,拷贝到画生成物的窗口中,再将结构调整为产物就可以了。

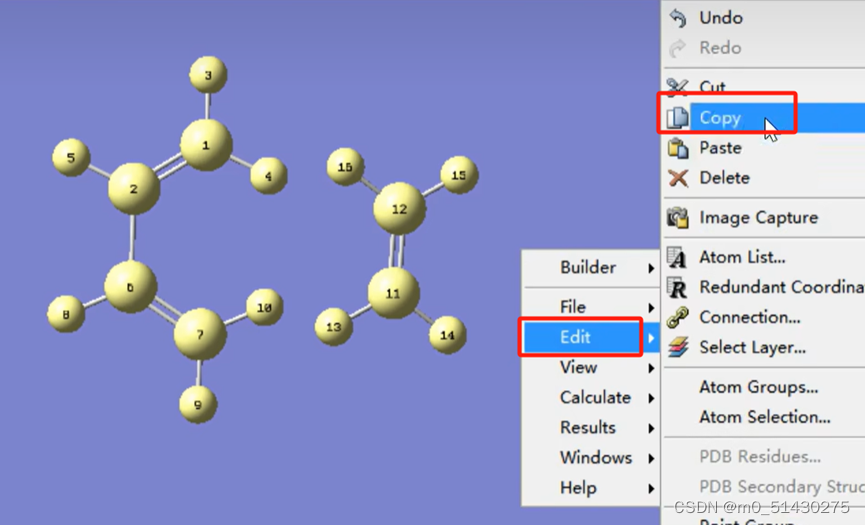
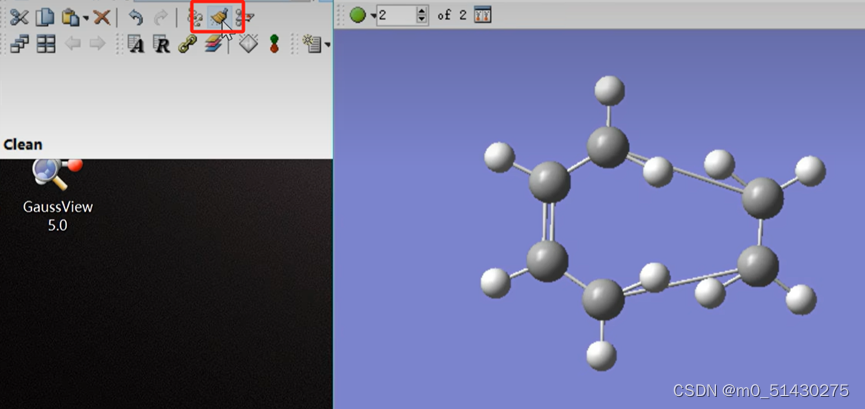
如下图所示:画好大概的生成物,连上该连的键,然后点击clean,让软件帮忙调整:
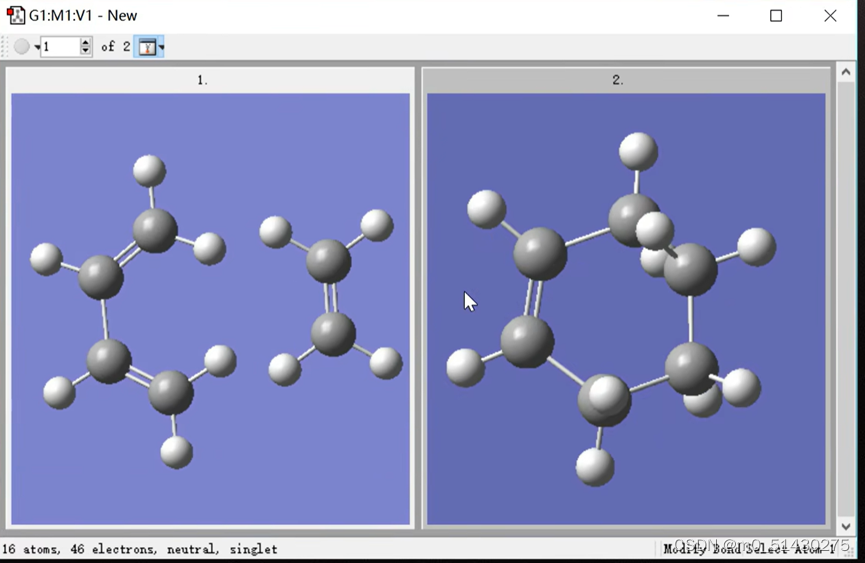
下面是画好反应物与生成物的结果:
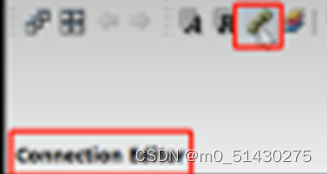
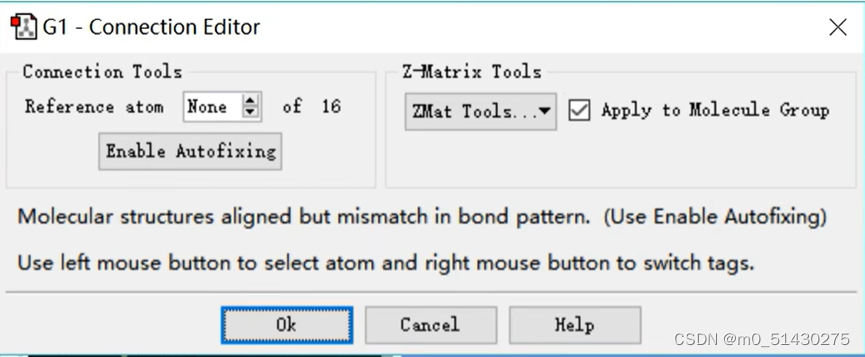
找到Connection Editor。它可以在多个分子中指定加原子,或者说在分子集合中判断原子的排列顺序是否合理,从而比较和编辑分子自动排列原子。
1.可以自动或者手动来编制分子中各个原子的序号
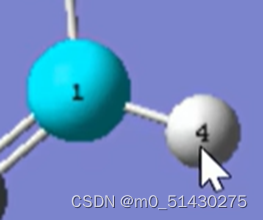
以上图为例,在反应物中,当左键点击1号C原子,右键点击4号H原子之后,二者序号互换。但在生成物中它不会有变化,如下图所示:

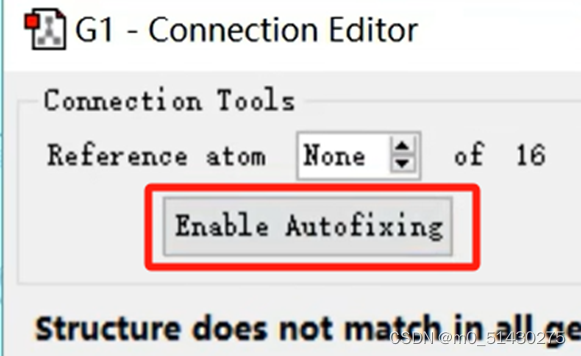
如果原子很多,手动调整就不方便。当反应物与生成物原子序号不一致时,点击Enable Autofixing可以自动编号:
右边功能:

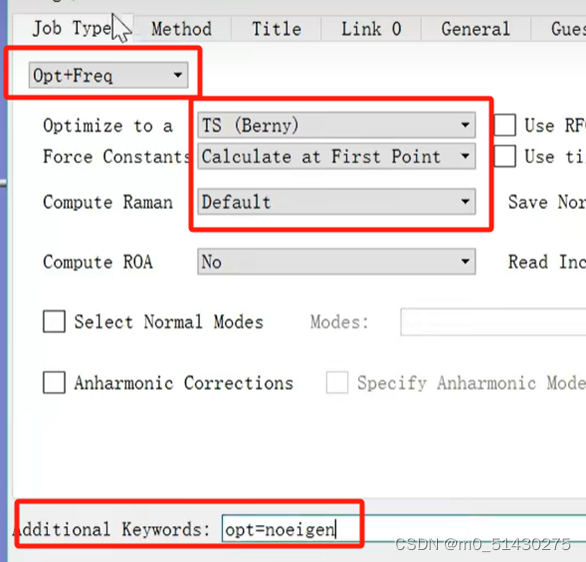
在过渡态计算中需要选择opt+freq,参数和关键词添加(additional keyword)也要改,如下图所示:
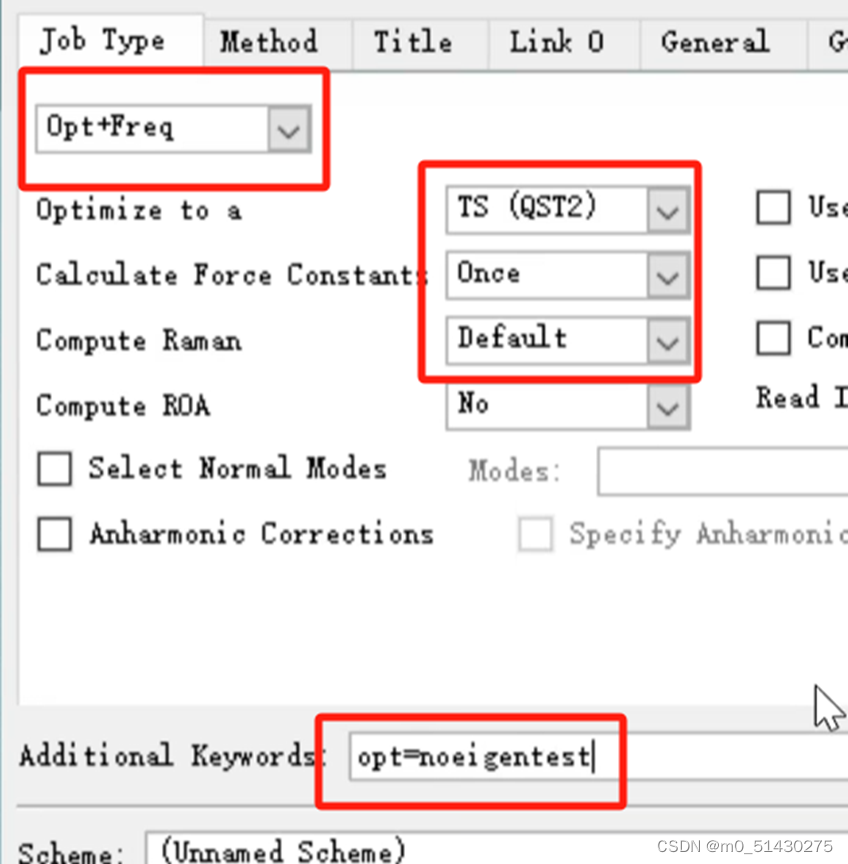
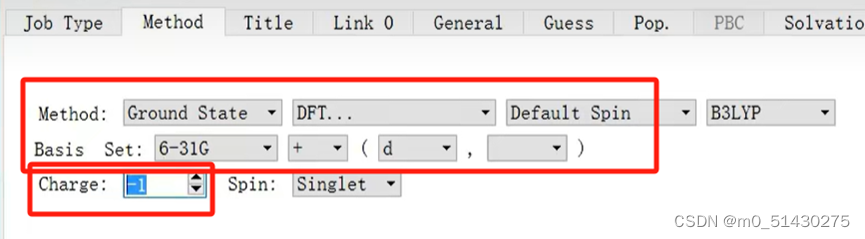
然后再对它进行一个方法和基组的选择:
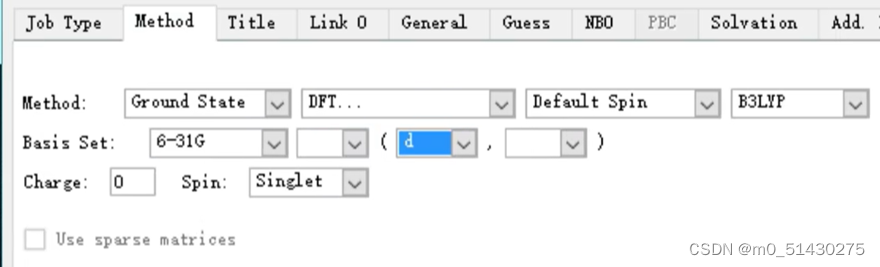
设置link0选项的参数:
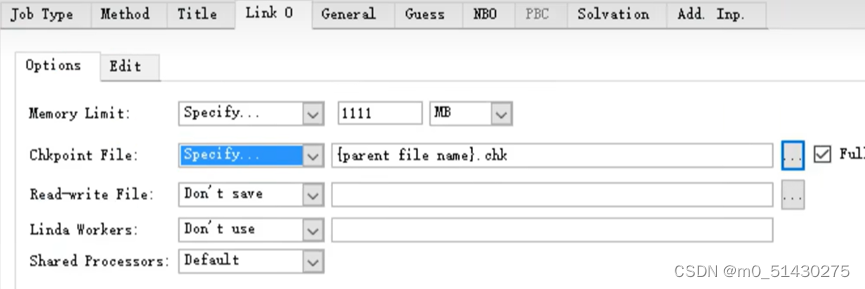
跑出结果如图所示:
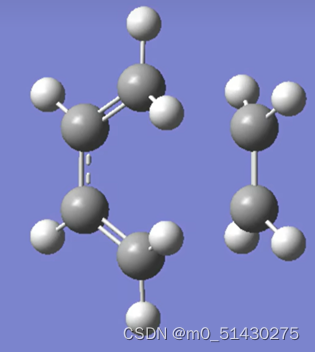
点击Result-Vibrations:
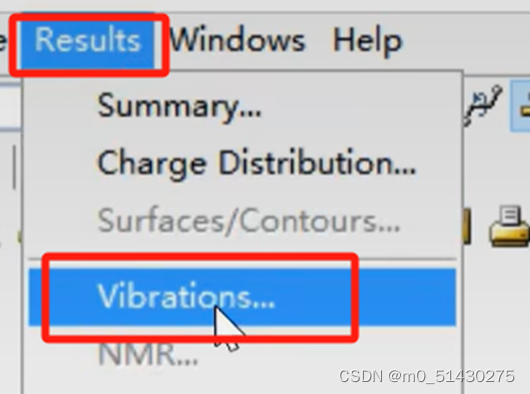
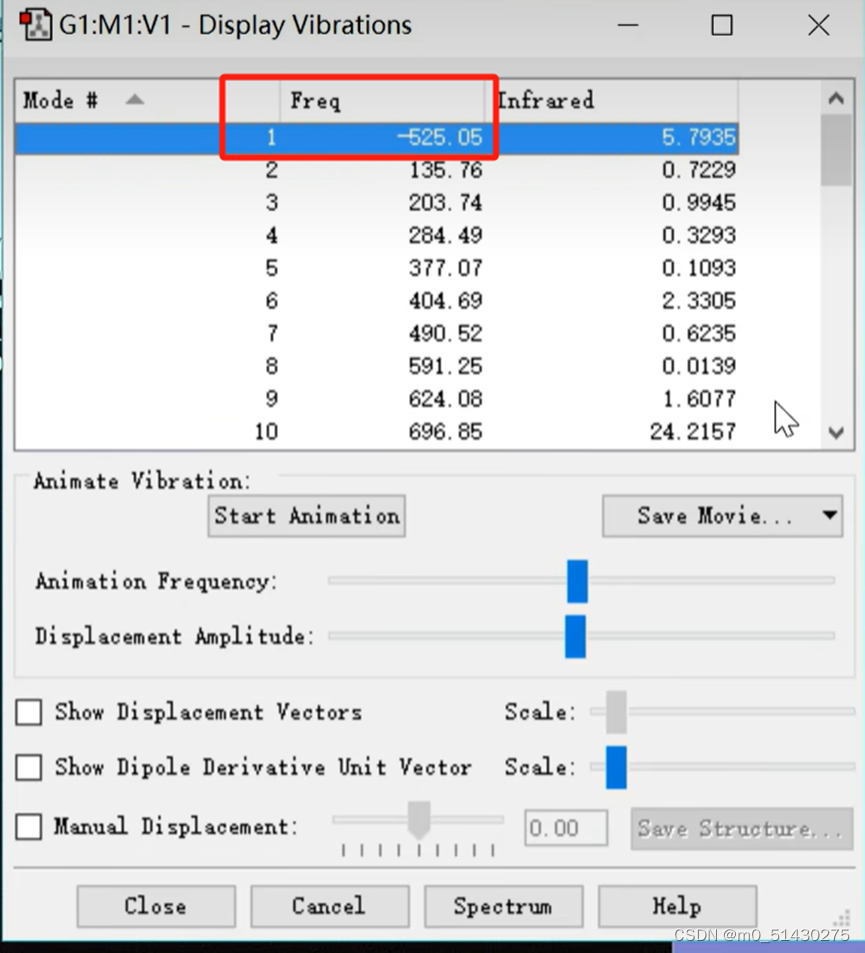
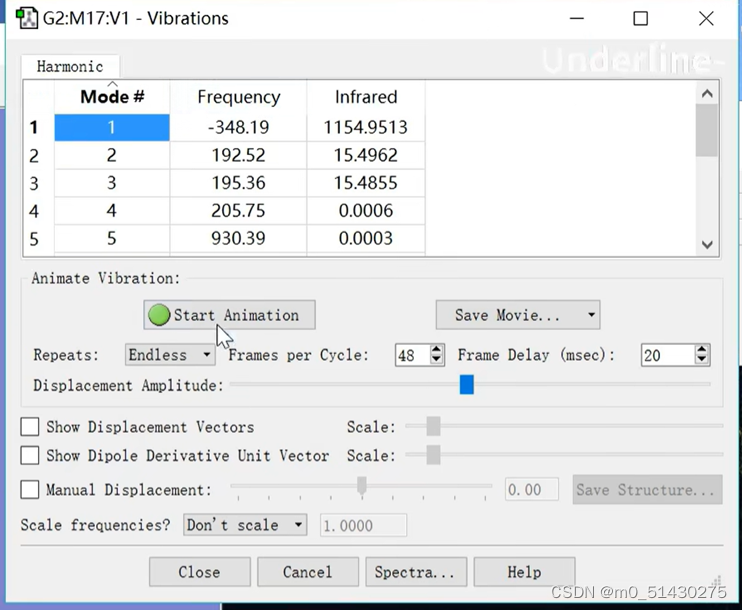
可以看到有且仅有一个虚频,这表示我们找到了一个过渡态:
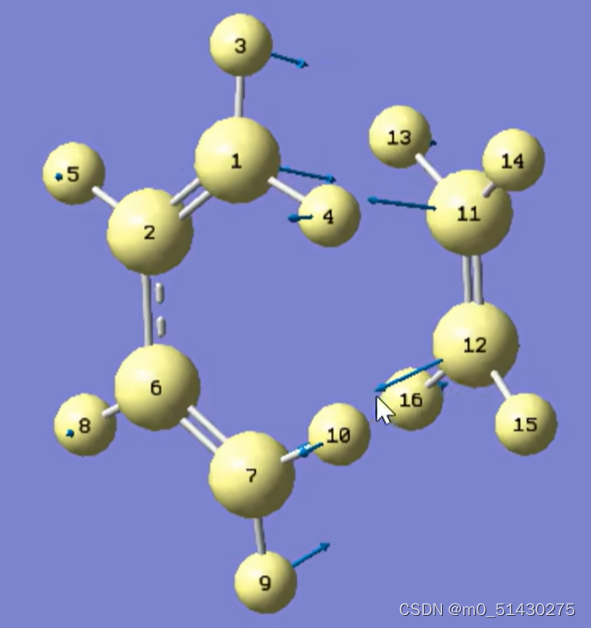
点击Start Animation,可以看到震动趋势。这也反映了反应时原子进攻的方向:
TS预测过渡态
TS方法需要猜测过渡态结构,以下图为例,中间的那个物质为猜测的过渡态:
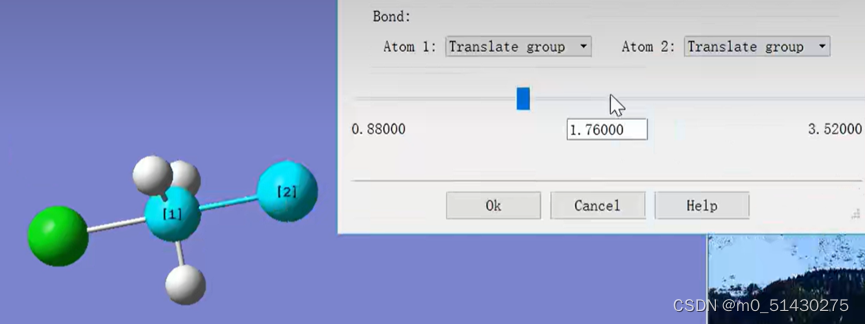
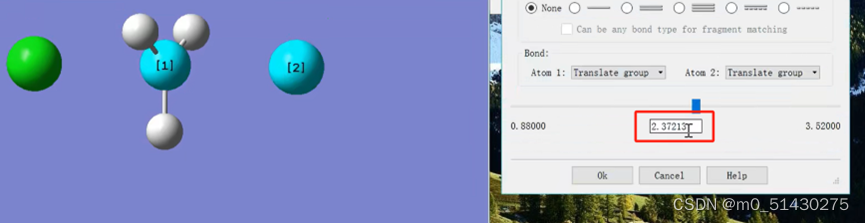
画出猜测的过渡态的结构。查看Cl原子的键长,在过渡态猜测的时候,根据经验一般都会将涉及到反应的键的键长提高30%,从而更加接近合理的过渡态结构。因此将1.76改为2.88
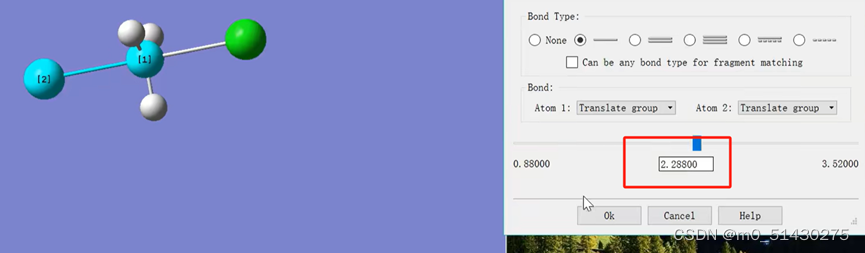
接下来就可以开始递交计算工作,参数设置如下图所示:
将设置文件保存后,打开gaussian,载入刚刚保存的.gjf文件,点击计算:

接着在gauss view 打开计算结果.out文件。打开键长工具,看到键长为2.371。与我们一开始猜测的比较接近。
接着又打开Vibrations,查看震动方向和频率:

震动方向与实际反应一致,并且可以看到只有一个虚频,因此基本可以确定猜测的过渡态结构就是我们需要寻找的过渡态:
IRC(反应路径跟踪)的计算
对于一个过渡态,它在一个方向上是极大值点,那么在此方向上往前或往后走都能找到最小值点,对应的物理意义就是通过过渡态推出了反应的起始物与产物。IRC计算就是这样的一个过程,先通过过渡态计算获得准确的过渡态结构,然后跑IRC即可获得反应路径。
下图是之前通过QST2方法得到的过渡态,需要注意的是,初始的结构必须是已经优化好了的过渡态结构;设定的计算参数一定要和过渡态计算时候设定的一模一样,不能使用其他的方法或者基组

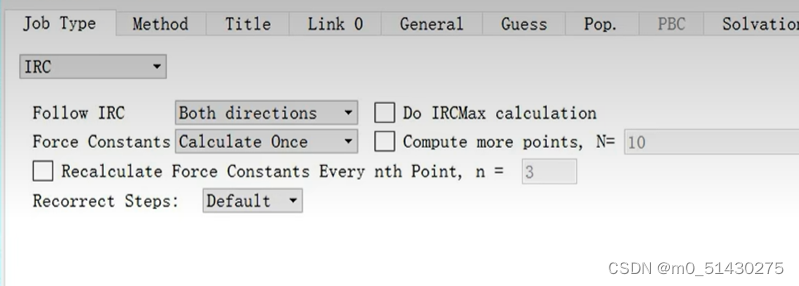
如下图所示,计算的参数要和之前选择的一样:
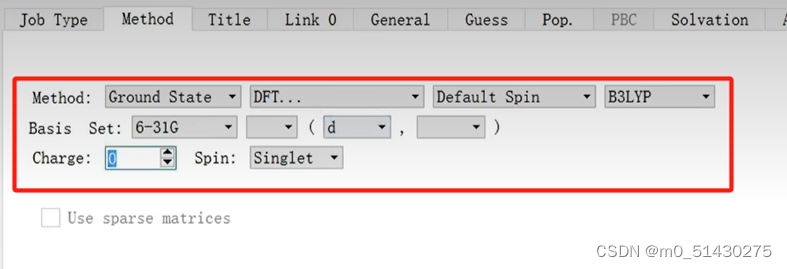
在additional keyword里面添加参数,Maxpoints规定了向两边搜索极小值点允许走的最大步数,Stepsize规定了步长。前者会影响耗时,后者会影响精确度。
默认算法改为LQA,可以解决一些因为不收敛而报错的问题。
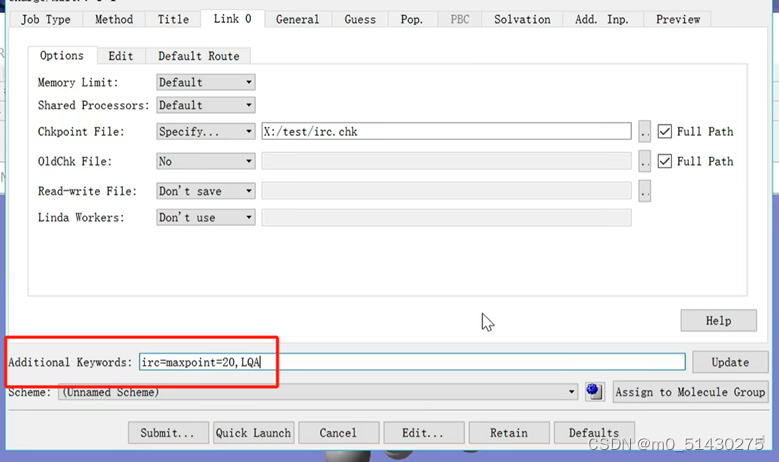
保存文件为gjf格式,然后用gaussian对它进行计算,计算的结果用gauss view打开:

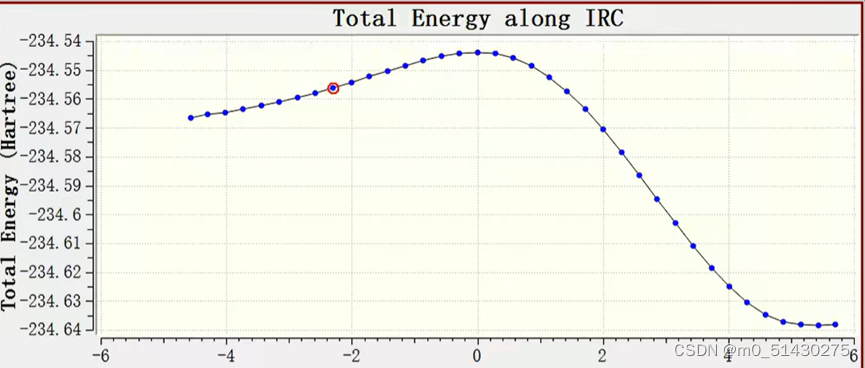
可以看到IRC路径能量变化,一个一个移动光标选择的点的位置可以在gauss view窗口看到过渡态结构的变化,两边端点就是对应反应物(图二)和生成物(图三):
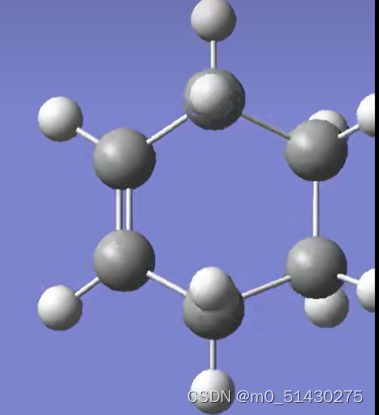
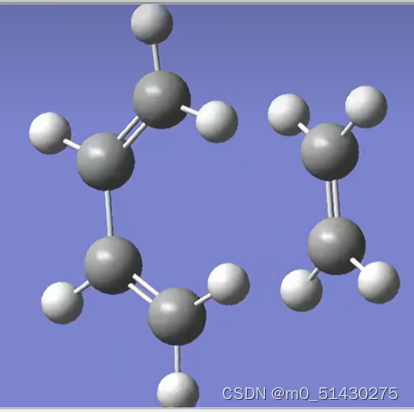
如果想看一些键长键角的变化,例如看看4与5号C原子键长变化:
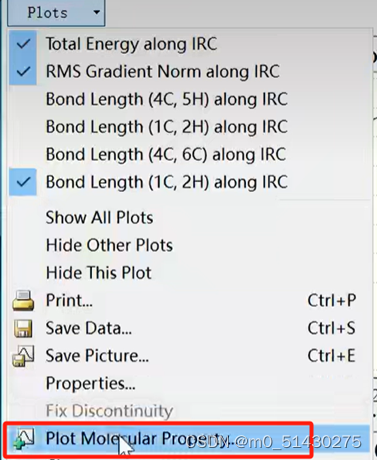

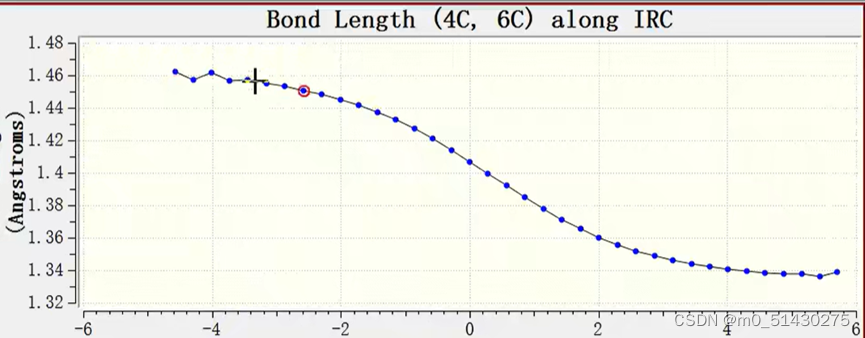
在.out文件可以查看各种参数:

Pathnumber=1 代表正方向,2代表反方向:

用chem3D预优化chemdraw画出来的平面式
将.mo;文件拖入chem3D中:
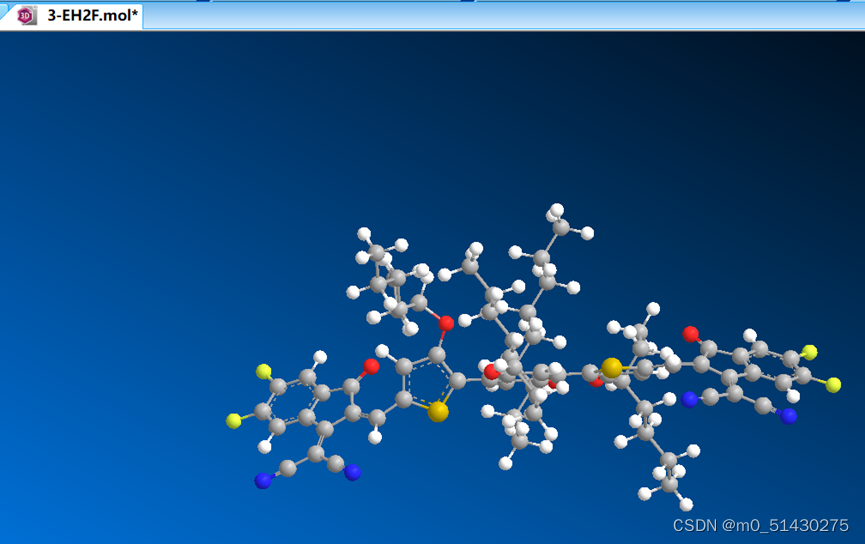
然后点击caculations,下图所示:
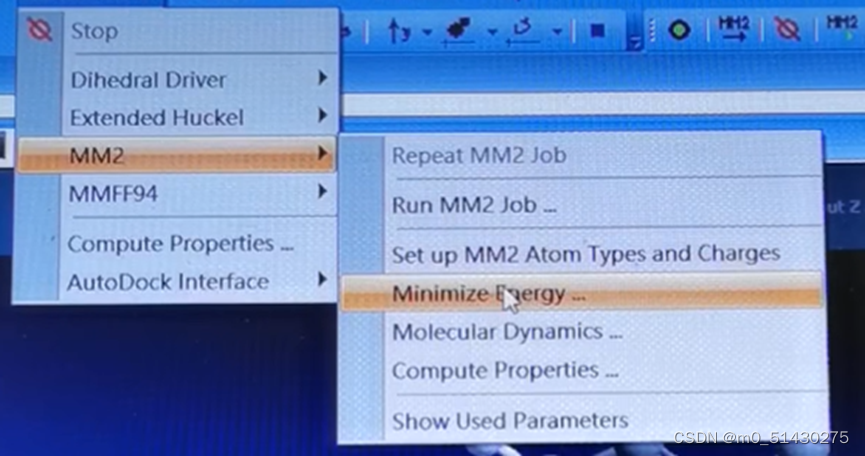
跑出来结果如图所示:
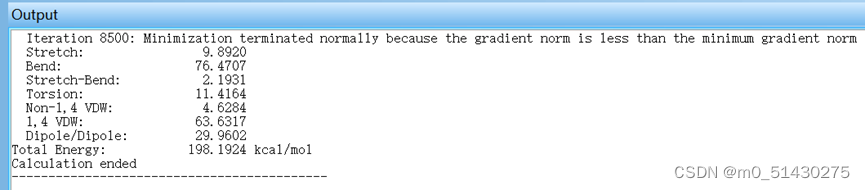
可以将文件保存为.out文件或者.pbd文件。
















 2470
2470











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









