







Abstract
药物推荐是医疗保健领域人工智能的一项重要任务。现有工作仅基于患者的电子健康记录,专注于为患有复杂健康状况的患者推荐药物组合。因此,它们存在以下局限性:
- 一些重要数据,如药物分子结构,在推荐过程中未被利用。
- 药物 - 药物相互作用(DDI)是隐式建模的,这可能导致次优结果。
为解决这些局限性,我们提出了一种名为SafeDrug的DDI可控药物推荐模型,以利用药物的分子结构并显式地对DDI进行建模。SafeDrug配备了全局消息传递神经网络(MPNN)模块和局部二分学习模块,以充分编码药物分子的连通性和功能性。SafeDrug还具有可控损失函数,可有效控制推荐药物组合中的DDI水平。在一个基准数据集上,我们的SafeDrug相对而言显示出将DDI降低了19.43%,并且与之前的方法相比,推荐药物组合与实际开具药物组合之间的Jaccard相似度提高了2.88%。此外,SafeDrug所需的参数比之前基于深度学习的方法少得多,导致训练速度快约14%,推理速度快约2倍。
1 Introduction
如今,丰富的健康数据,如纵向电子健康记录(EHR)以及网络上可用的大量生物医学数据,使研究人员和医生能够为临床决策构建更好的预测模型[Choi等人,2016a;Xiao等人,2018]。其中,推荐有效且安全的药物组合是一项重要任务,特别是对于帮助患有复杂健康状况的患者[Shang等人,2019;Shang等人,2018]。推荐药物组合的主要目标是根据患者的健康状况为特定患者定制安全的药物组合。早期的药物推荐模型是基于实例的,仅基于当前的医院就诊情况[Zhang等人,2017;Wang等人,2017]。因此,新诊断为高血压的患者很可能与患有慢性未控制高血压的另一位患者得到相同的治疗。这种局限性影响了推荐的安全性和实用性。为克服这一问题,提出了诸如[Le等人,2018;Shang等人,2019]等纵向方法。它们利用纵向患者病史中的时间依赖性来提供更个性化的推荐。然而,它们仍然存在以下局限性:
- 药物编码不充分:现有工作[Zhang等人,2017;Shang等人,2019]通常使用独热编码来表示药物。每种药物被视为一个(二进制)单元,忽略了药物在其有意义的分子图表示中编码了重要的药物特性,如疗效和安全性概况。此外,分子子结构与功能相关。这些知识可用于提高药物推荐的准确性和安全性。
- 药物 - 药物相互作用(DDI)建模隐式且不可控:一些现有工作通过软约束或间接约束来建模药物 - 药物相互作用(DDI),如知识图谱(KGs)[Wang等人,2017;Mao等人,2019]和强化后处理[Zhang等人,2017]。这些对DDI的隐式处理导致最终推荐中不可控的比率或次优的推荐准确性。
为解决这些问题,我们提出了一种DDI可控的药物推荐模型,名为SafeDrug,以利用药物的分子结构并显式地对DDI进行建模。我们认为在药物推荐中纳入分子结构是有益的。我们的SafeDrug具有以下贡献:
- 双分子编码器捕获全局和局部分子模式:SafeDrug模型首先学习患者表示,将其输入到双分子编码器中,以捕获药物的全局药理特性[Brown和Fraser,1868]和局部子结构模式[Huang等人,2020b]。在全局方面,构建了一个消息传递神经网络(MPNN)编码器,逐层为整个药物层传递分子信息消息。药物的连通性信息在多跳中得到了很好的捕获。在局部方面,二分编码器将药物分子分割成子结构,每个子结构可能与小的功能基团相关联。在这项工作中,子结构表示被输入到一个有效的掩码神经网络中。模型的最终输出是通过全局和局部编码嵌入的元素级集成获得的。
- DDI可控损失函数:受比例 - 积分 - 微分(PID)控制器[Astrom和Hagglund,1995]的启发,我们设计了一种新技术,以相当大的灵活性自适应地结合监督损失和无监督DDI约束。在训练过程中,如果单个样本的DDI比率高于某个阈值/目标,则会强调并反向传播负DDI信号。在实验中,自适应梯度下降可以平衡模型准确性和最终DDI水平。通过预设目标,SafeDrug模型可以提供可靠的药物组合,以满足不同水平的DDI要求。
- 综合评估:我们遵循先前的工作[Zhang等人,2017;Shang等人,2019;Le等人,2018],并在从MIMIC - III数据库[Johnson等人,2016]提取的基准药物推荐数据集上评估SafeDrug。SafeDrug相对优于最佳基线,在DDI降低方面提高了19.43%,在Jaccard相似度方面提高了2.88%,在F1度量方面提高了2.14%。此外,与之前基于深度学习的药物推荐方法相比,SafeDrug需要的参数少得多,训练时间减少了14%,推理速度快约2倍。
数据处理文件和代码在Github上发布,论文的完整版本可在arXiv上找到。https://github.com/ycq091044/SafeDrug
2 Related Works
2.1 Medication Recommendation
现有的药物推荐算法可分为基于实例的和纵向推荐方法。基于实例的方法侧重于患者当前的健康状况。例如,LEAP[Zhang等人,2017]从当前就诊中提取特征信息,并采用多实例多标签药物推荐设置。提出纵向方法是为了利用临床病史中的时间依赖性,参见[Choi等人,2016a;Xiao等人,2018]。其中,RETAIN[Choi等人,2016b]利用具有反向时间注意力的两级RNN来建模纵向信息。GAMENet[Shang等人,2019]采用记忆神经网络,并将历史药物信息存储为进一步预测的参考。这些方法要么将最终药物组合建模为多标签二分类[Choi等人,2016b;Shang等人,2019;Shang等人,2018],要么通过顺序决策制定[Zhang等人,2017]。尽管它们取得了初步成功,但这些现有工作仍然存在以下局限性。其他重要数据,如药物分子结构,可以帮助增强药物推荐,但现有模型尚未利用。此外,现有工作对DDI的建模不充分,要么通过软约束,要么通过间接约束。
本文提出了一种DDI可控的药物推荐模型,该模型利用药物的分子结构并有效地对DDI进行建模。
2.2 Molecule Representation
由于分子结构与性质(如疗效和安全性)之间的关联,分子图表示仍然是一个重要的话题。早期,分子描述符[Mauri等人,2006]和药物指纹[Rogers和Hahn,2010;Duvenaud等人,2015]通常用于表示药物分子。近年来开发了深度学习模型来学习分子表示和建模分子子结构(一组相连的原子)。例如,[Huang等人,2020b]在SMILES字符串上开发了一种序列模式挖掘方法,并将一对药物建模为它们的子串表示的集合。此外,[Huang等人,2020a]提出使用基于图的神经网络模型直接建模分子图。
在本文中,受[Huang等人,2020a]的启发,提出SafeDrug在推荐过程中捕获分子的全局和局部信息。
3 Problem Formulation
电子健康记录(EHR)
患者的电子健康记录数据以医疗代码(例如,诊断、程序和药物)的纵向向量形式捕获患者的综合病史。形式上,患者 j j j的电子健康记录可以表示为序列 X j = [ x j ( 1 ) , x j ( 2 ) , … , x j ( V j ) ] X_j = [x_j^{(1)}, x_j^{(2)}, \ldots, x_j^{(V_j)}] Xj=[xj(1),xj(2),…,xj(Vj)],其中 V j V_j Vj是患者 j j j的就诊总次数。 X j X_j Xj的第 i i i个条目记录了患者 j j j的第 i i i次就诊,由 x j ( i ) = [ d j ( i ) , p j ( i ) , m j ( i ) ] x_j^{(i)} = [d_j^{(i)}, p_j^{(i)}, m_j^{(i)}] xj(i)=[dj(i),pj(i),mj(i)]指定,其中 d j ( i ) ∈ { 0 , 1 } ∣ D ∣ d_j^{(i)} \in \{0, 1\}^{|\mathcal{D}|} dj(i)∈{0,1}∣D∣, p j ( i ) ∈ { 0 , 1 } ∣ P ∣ p_j^{(i)} \in \{0, 1\}^{|\mathcal{P}|} pj(i)∈{0,1}∣P∣, m j ( i ) ∈ { 0 , 1 } ∣ M ∣ m_j^{(i)} \in \{0, 1\}^{|\mathcal{M}|} mj(i)∈{0,1}∣M∣,分别是多热诊断、程序和药物向量。 D \mathcal{D} D、 P \mathcal{P} P、 M \mathcal{M} M是相应的元素集, ∣ ⋅ ∣ |\cdot| ∣⋅∣表示基数。
安全药物组合推荐
我们专注于药物推荐,同时降低/控制预测中的药物 - 药物相互作用(DDI)率。我们使用对称二进制邻接矩阵
D
∈
{
0
,
1
}
∣
M
∣
×
∣
M
∣
D \in \{0, 1\}^{|\mathcal{M}| \times |\mathcal{M}|}
D∈{0,1}∣M∣×∣M∣来表示DDI关系。
D
i
j
=
1
D_{ij} = 1
Dij=1表示药物
i
i
i和
j
j
j之间的相互作用已被报道,而
D
i
j
=
0
D_{ij} = 0
Dij=0表示安全的联合处方。本文旨在学习一个药物推荐函数
f
(
⋅
)
f(\cdot)
f(⋅),使得对于每次就诊,例如第
t
t
t次就诊,给定患者到目前为止的诊断和程序序列
[
d
(
1
)
,
d
(
2
)
,
…
,
d
(
t
)
]
[d^{(1)}, d^{(2)}, \ldots, d^{(t)}]
[d(1),d(2),…,d(t)]和
[
p
(
1
)
,
p
(
2
)
,
…
,
p
(
t
)
]
[p^{(1)}, p^{(2)}, \ldots, p^{(t)}]
[p(1),p(2),…,p(t)],以及DDI矩阵
D
D
D,
f
(
⋅
)
f(\cdot)
f(⋅)可以生成药物推荐:
m
^
(
t
)
=
f
(
[
d
(
i
)
]
i
=
1
t
,
[
p
(
i
)
]
i
=
1
t
)
∈
{
0
,
1
}
∣
M
∣
.
(
1
)
\hat{m}^{(t)} = f ([d^{(i)}]_{i = 1}^{t}, [p^{(i)}]_{i = 1}^{t}) \in \{0, 1\}^{|\mathcal{M}|}. \quad (1)
m^(t)=f([d(i)]i=1t,[p(i)]i=1t)∈{0,1}∣M∣.(1)
目标函数由两部分组成:(i)提取真实药物组合
m
(
t
)
m^{(t)}
m(t),作为对
m
^
(
t
)
\hat{m}^{(t)}
m^(t)的监督惩罚;(ii)使用矩阵
D
D
D推导对
m
^
(
t
)
\hat{m}^{(t)}
m^(t)的无监督DDI约束。
4 The SafeDrug Model
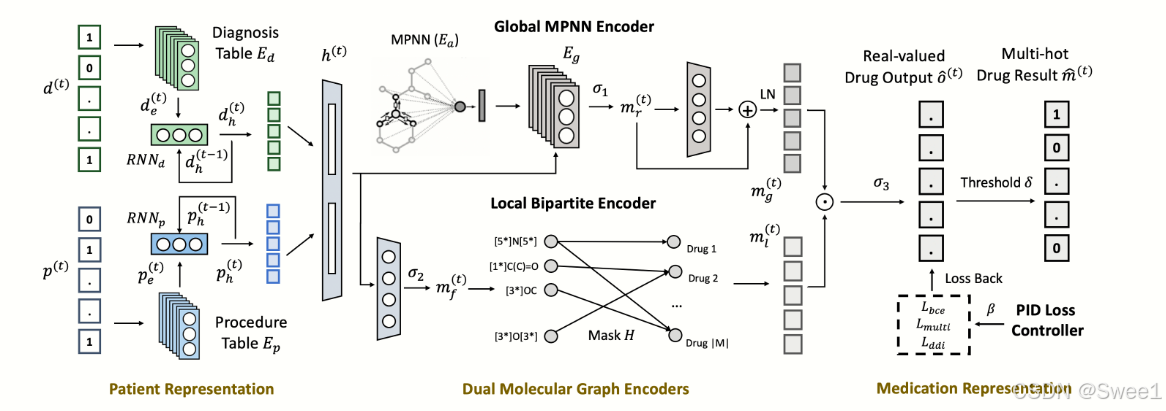
图1:SafeDrug模型。我们首先通过RNN(循环神经网络)对诊断和程序序列进行编码,以生成患者健康表示
h
(
t
)
h^{(t)}
h(t);然后,该表示通过双分子图编码器,以获得全局和局部分子结构嵌入
m
g
(
t
)
m_g^{(t)}
mg(t)和
m
l
(
t
)
m_l^{(t)}
ml(t);最后将这两个嵌入向量组合并进行阈值处理,以获得推荐的药物组合
m
^
(
t
)
\hat{m}^{(t)}
m^(t)。
如图1所示,我们的SafeDrug模型由四个组件组成:
- 纵向患者表示模块:从患者的电子健康记录(EHR)数据中学习患者表示。
- 全局消息传递神经网络(MPNN)编码器:将患者表示作为输入,并输出一个全局药物向量,其中每个条目量化患者表示与每个药物表示之间的相似性。
- 二分编码器(并行):也采用相同的患者表示,并输出一个局域药物向量,该向量对药物的分子子结构功能进行编码。
- 药物表示模块:最后,全局和局域药物表示向量在该模块中按元素方式组合,最终的药物输出是通过按元素阈值处理获得的。
4.1 Longitudinal Patient Representation
从纵向电子健康记录(EHR)数据中,患者健康可以通过他们的诊断和程序信息进行编码。
诊断嵌入
为了充分利用丰富的诊断信息,我们设计了一个嵌入表
E
d
∈
R
∣
D
∣
×
d
i
m
E_d \in \mathbb{R}^{|\mathcal{D}| \times dim}
Ed∈R∣D∣×dim,其中每一行存储特定诊断的嵌入向量,
d
i
m
dim
dim是嵌入空间的维度。给定一个多热诊断向量
d
(
t
)
∈
{
0
,
1
}
∣
D
∣
d^{(t)} \in \{0, 1\}^{|\mathcal{D}|}
d(t)∈{0,1}∣D∣,我们通过向量 - 矩阵点积将相应的诊断代码投影到嵌入空间中,这本质上是对每个诊断嵌入进行求和,即:
d
e
(
t
)
=
d
(
t
)
E
d
.
(
2
)
d_e^{(t)} = d^{(t)}E_d. \quad (2)
de(t)=d(t)Ed.(2)
在训练期间,
E
d
E_d
Ed是可学习的,并且在每次就诊和每个患者之间共享。
程序嵌入
类似地,还设计了一个共享的程序表
E
p
∈
R
∣
P
∣
×
d
i
m
E_p \in \mathbb{R}^{|\mathcal{P}| \times dim}
Ep∈R∣P∣×dim来编码相关的程序向量
p
(
t
)
p^{(t)}
p(t)(也是一个多热向量),即:
p
e
(
t
)
=
p
(
t
)
E
p
.
(
3
)
p_e^{(t)} = p^{(t)}E_p. \quad (3)
pe(t)=p(t)Ep.(3)
两个嵌入向量
d
e
(
t
)
,
p
e
(
t
)
∈
R
d
i
m
d_e^{(t)}, p_e^{(t)} \in \mathbb{R}^{dim}
de(t),pe(t)∈Rdim,编码了患者当前的健康状况。实际上,一个健康快照可能不足以做出治疗决策。例如,为一位新诊断为高血压的患者和另一位患有慢性高血压超过两年的患者开具相同的药物是不合适的。
动态患者病史建模
这里我们使用RNN(循环神经网络)模型来建模动态患者病史。具体来说,我们使用两个单独的RNN来获得隐藏的诊断和程序向量
d
h
(
t
)
d_h^{(t)}
dh(t)和
p
h
(
t
)
p_h^{(t)}
ph(t),以防其中一个序列在实践中可能无法获取,即:
d
h
(
t
)
=
R
N
N
d
(
d
e
(
t
)
,
d
h
(
t
−
1
)
)
=
R
N
N
d
(
d
e
(
t
)
,
…
,
d
e
(
1
)
)
,
(
4
)
d_h^{(t)} = RNN_d (d_e^{(t)}, d_h^{(t - 1)}) = RNN_d (d_e^{(t)}, \ldots, d_e^{(1)}), \quad (4)
dh(t)=RNNd(de(t),dh(t−1))=RNNd(de(t),…,de(1)),(4)
p
h
(
t
)
=
R
N
N
p
(
p
e
(
t
)
,
p
h
(
t
−
1
)
)
=
R
N
N
p
(
p
e
(
t
)
,
…
,
p
e
(
1
)
)
,
(
5
)
p_h^{(t)} = RNN_p (p_e^{(t)}, p_h^{(t - 1)}) = RNN_p (p_e^{(t)}, \ldots, p_e^{(1)}), \quad (5)
ph(t)=RNNp(pe(t),ph(t−1))=RNNp(pe(t),…,pe(1)),(5)
其中
d
h
(
t
)
,
p
h
(
t
)
∈
R
d
i
m
d_h^{(t)}, p_h^{(t)} \in \mathbb{R}^{dim}
dh(t),ph(t)∈Rdim。并且
d
h
(
0
)
,
p
h
(
0
)
d_h^{(0)}, p_h^{(0)}
dh(0),ph(0)是全零向量。
患者表示
然后我们将诊断嵌入
d
h
(
t
)
d_h^{(t)}
dh(t)和程序嵌入
p
h
(
t
)
p_h^{(t)}
ph(t)连接成一个更紧凑的患者表示
h
(
t
)
h^{(t)}
h(t)。我们遵循一种常见且有效的方法,首先将两个向量连接成一个双倍长的向量,然后应用一个前馈神经网络
N
N
1
(
⋅
)
:
R
2
d
i
m
↦
R
d
i
m
NN_1(\cdot) : \mathbb{R}^{2dim} \mapsto \mathbb{R}^{dim}
NN1(⋅):R2dim↦Rdim(
W
1
W_1
W1是参数,
#
\#
#是连接操作),即:
h
(
t
)
=
N
N
1
(
[
d
h
(
t
)
#
p
h
(
t
)
]
;
W
1
)
.
(
6
)
h^{(t)} = NN_1([d_h^{(t)} \# p_h^{(t)}]; W_1). \quad (6)
h(t)=NN1([dh(t)#ph(t)];W1).(6)
至此,我们获得了一个整体的患者表示
h
(
t
)
∈
R
d
i
m
h^{(t)} \in \mathbb{R}^{dim}
h(t)∈Rdim,接下来我们通过全面建模药物分子数据库,基于此生成安全的药物推荐。
4.2 Dual Molecular Graph Encoders
为了研究药物性质及其依赖性,我们利用全局分子连通性特征和局部分子功能性。在以下部分,我们并行介绍两个新的分子图编码器,以全面编码药物分子。
(I)全局MPNN编码器
我们使用具有可学习指纹的消息传递神经网络(MPNN)算子对药物分子数据进行编码,旨在将单个分子图中的原子信息卷积和池化为向量表示。
首先,我们收集所有出现的原子集合 C = { a i } \mathcal{C} = \{a_i\} C={ai},并设计一个可学习的原子嵌入表 E a ∈ R ∣ C ∣ × d i m E_a \in \mathbb{R}^{|\mathcal{C}| \times dim} Ea∈R∣C∣×dim,其中每一行查找特定原子的初始嵌入/指纹。我们的MPNN模型设计与先前的工作[Coley等人,2017;Yang等人,2019]非常不同,这些工作大多使用原子描述符,例如原子质量和手性,作为初始特征。然而,对于分子结构建模,原子 - 原子连通性比单个原子更重要。
给定一个药物分子图(原子
a
0
,
…
,
a
n
a_0, \ldots, a_n
a0,…,an作为顶点,原子 - 原子键作为边),其邻接矩阵为
A
\mathbf{A}
A,以及来自
E
a
E_a
Ea的初始原子指纹,即
{
y
0
(
0
)
,
y
1
(
0
)
,
…
,
y
n
(
0
)
}
\{y_0^{(0)}, y_1^{(0)}, \ldots, y_n^{(0)}\}
{y0(0),y1(0),…,yn(0)},我们将图上的逐层消息传递公式化为:
z
i
(
l
+
1
)
=
∑
j
:
A
i
j
=
1
MESSAGE
l
(
y
i
(
l
)
,
y
j
(
l
)
;
W
(
l
)
)
,
(
7
)
z_i^{(l + 1)} = \sum_{j: \mathbf{A}_{ij} = 1} \text{MESSAGE}_l (y_i^{(l)}, y_j^{(l)}; \mathbf{W}^{(l)}), \quad (7)
zi(l+1)=j:Aij=1∑MESSAGEl(yi(l),yj(l);W(l)),(7)
y
i
(
l
+
1
)
=
UPDATE
l
(
y
i
(
l
)
,
z
i
(
l
+
1
)
)
,
i
=
0
,
…
,
n
(
8
)
y_i^{(l + 1)} = \text{UPDATE}_l (y_i^{(l)}, z_i^{(l + 1)}), i = 0, \ldots, n \quad (8)
yi(l+1)=UPDATEl(yi(l),zi(l+1)),i=0,…,n(8)
其中
l
l
l是层索引,
z
i
(
l
+
1
)
z_i^{(l + 1)}
zi(l+1)是在第
l
l
l次迭代时来自原子
i
i
i的邻居的编码消息,
y
i
(
l
+
1
)
y_i^{(l + 1)}
yi(l+1)是原子
i
i
i的隐藏状态。在消息传递过程中,
z
i
(
l
+
1
)
z_i^{(l + 1)}
zi(l+1)和
y
i
(
l
+
1
)
y_i^{(l + 1)}
yi(l+1)与每个顶点原子
i
i
i一起使用消息函数
MESSAGE
l
(
⋅
)
\text{MESSAGE}_l(\cdot)
MESSAGEl(⋅)和顶点更新函数
UPDATE
l
(
⋅
)
\text{UPDATE}_l(\cdot)
UPDATEl(⋅)进行更新,并且
W
(
l
)
\mathbf{W}^{(l)}
W(l)是逐层参数矩阵。在应用
L
L
L层消息传递后,药物分子的全局表示通过读出函数进行池化,该函数计算所有原子指纹的平均值,即:
y
=
READOUT
(
{
y
i
(
L
)
∣
i
=
0
,
…
,
n
}
)
.
(
9
)
y = \text{READOUT} (\{y_i^{(L)} | i = 0, \ldots, n\}). \quad (9)
y=READOUT({yi(L)∣i=0,…,n}).(9)
我们对每个单个药物分子应用具有共享参数的相同MPNN编码器(总共有
∣
M
∣
|\mathcal{M}|
∣M∣种不同的药物)。我们将所有药物分子的MPNN嵌入收集到药物记忆
E
g
∈
R
∣
M
∣
×
d
i
m
E_g \in \mathbb{R}^{|\mathcal{M}| \times dim}
Eg∈R∣M∣×dim中,其中每一行是一种药物的嵌入。
患者与药物匹配当将患者表示
h
(
t
)
h^{(t)}
h(t)视为查询时,我们旨在从记忆
E
g
E_g
Eg中搜索最相关的药物。健康与药物的匹配分数通过MPNN表示和患者表示
h
(
t
)
h^{(t)}
h(t)的点积计算。对于所有药物,我们将
E
g
E_g
Eg的每一行与
h
(
t
)
h^{(t)}
h(t)进行操作,然后通过一个sigmoid函数
σ
1
(
⋅
)
\sigma_1(\cdot)
σ1(⋅),即:
m
r
(
t
)
=
σ
1
(
E
g
h
(
t
)
)
.
(
10
)
m_r^{(t)} = \sigma_1 (E_g h^{(t)}). \quad (10)
mr(t)=σ1(Egh(t)).(10)
m
r
(
t
)
m_r^{(t)}
mr(t)的每个元素存储一种药物的匹配分数。鉴于Transformer[Vaswani等人,2017]中最近成功的后LN架构[Wang等人,2019],匹配分数进一步通过一个前馈神经网络
N
N
2
(
⋅
)
:
R
d
i
m
↦
R
d
i
m
NN_2(\cdot) : \mathbb{R}^{dim} \mapsto \mathbb{R}^{dim}
NN2(⋅):Rdim↦Rdim进行参数化,然后进行带有残差连接的层归一化(LN)步骤,即:
m
g
(
t
)
=
L
N
(
m
r
(
t
)
+
N
N
2
(
m
r
(
t
)
;
W
2
)
)
.
(
11
)
m_g^{(t)} = LN (m_r^{(t)} + NN_2 (m_r^{(t)}; W_2)). \quad (11)
mg(t)=LN(mr(t)+NN2(mr(t);W2)).(11)
(II)局部分割编码器
使用MPNN编码器,具有相似结构的分子应被映射到附近的空间。然而,在结构域上有显著重叠的一对药物在药物 - 药物相互作用(DDI)或其他功能活性方面可能表现不同[Ryu等人,2018]。实际上,药物的功能更好地由分子子结构反映,这些子结构被定义为一组可能具有相关官能团的相连原子。本节重点关注分子的子结构组成,并构建一个子结构到药物的二分架构,以捕获药物的局部功能及其依赖性。
分子分割为了捕获药物分子与其子结构之间的关系,我们首先采用著名的分子分割方法,即反向合成有趣的化学子结构(BRICS)[Degen等人,2008],它保留了最关键的药物官能团和更重要的键。SafeDrug可以轻松支持其他分割方法,如RECAP、ECFP。分割后,每个分子被分解为一组化学子结构。在这种设置下,一对药物可能在一组共同的子结构上相交。我们将总体子结构集表示为 S \mathcal{S} S。基于许多关系,很容易构建一个由掩码矩阵 H ∈ { 0 , 1 } ∣ S ∣ × ∣ M ∣ \mathbf{H} \in \{0, 1\}^{|\mathcal{S}| \times |\mathcal{M}|} H∈{0,1}∣S∣×∣M∣表示的二分架构: H i j = 1 \mathbf{H}_{ij} = 1 Hij=1表示药物 j j j包含子结构 i i i。
二分学习我们希望
H
\mathbf{H}
H能够帮助在给定输入
h
(
t
)
h^{(t)}
h(t)的情况下,基于其功能信息推导出药物表示。为此,我们首先应用一个前馈神经网络
N
N
3
(
⋅
)
:
R
d
i
m
↦
R
∣
S
∣
NN_3(\cdot) : \mathbb{R}^{dim} \mapsto \mathbb{R}^{|\mathcal{S}|}
NN3(⋅):Rdim↦R∣S∣进行维度转换,然后通过一个sigmoid函数
σ
2
(
⋅
)
\sigma_2(\cdot)
σ2(⋅)。实际上,
N
N
3
(
⋅
)
NN_3(\cdot)
NN3(⋅)将患者表示
h
(
t
)
h^{(t)}
h(t)转换为局部功能向量,即:
m
f
(
t
)
=
σ
2
(
N
N
3
(
h
(
t
)
;
W
3
)
)
.
(
12
)
m_f^{(t)} = \sigma_2 (NN_3 (h^{(t)}; W_3)). \quad (12)
mf(t)=σ2(NN3(h(t);W3)).(12)
m
f
(
t
)
m_f^{(t)}
mf(t)的每个条目量化了相应功能的重要性。因此,
m
f
(
t
)
m_f^{(t)}
mf(t)可以被视为当前患者的抗病功能组合。接下来,我们旨在生成能够涵盖所有这些抗病功能同时考虑预防DDI的药物推荐。
因此,我们进行网络剪枝并设计一个掩码的1层神经网络
N
N
4
(
⋅
)
:
R
∣
S
∣
↦
R
∣
M
∣
NN_4(\cdot) : \mathbb{R}^{|\mathcal{S}|} \mapsto \mathbb{R}^{|\mathcal{M}|}
NN4(⋅):R∣S∣↦R∣M∣,其中参数矩阵通过二分架构
H
\mathbf{H}
H进行掩码,本质上是通过矩阵元素级乘积
⊙
\odot
⊙。在训练过程中,
N
N
4
(
⋅
)
NN_4(\cdot)
NN4(⋅)将学习将功能向量
m
f
(
t
)
m_f^{(t)}
mf(t)映射到相应的局域药物表示,即:
m
l
(
t
)
=
N
N
4
(
m
f
(
t
)
;
W
4
⊙
H
)
.
(
13
)
m_l^{(t)} = NN_4 (m_f^{(t)}; W_4 \odot \mathbf{H}). \quad (13)
ml(t)=NN4(mf(t);W4⊙H).(13)
我们表明掩码
H
\mathbf{H}
H将使模型能够(i)具有少得多的参数(由于
H
\mathbf{H}
H的稀疏性);(ii)通过避免相互作用药物的联合处方来预防DDI(我们在附录中证明了这一点)。
4.3 Medication Representation
最终的药物表示是通过将全局药物匹配向量视为注意力信号来进一步调整局域药物功能表示得到的。我们最终使用一个sigmoid函数
σ
3
(
⋅
)
\sigma_3(\cdot)
σ3(⋅)来缩放输出,即:
o
^
(
t
)
=
σ
3
(
m
g
(
t
)
⊙
m
l
(
t
)
)
,
(
14
)
\hat{o}^{(t)} = \sigma_3 (m_g^{(t)} \odot m_l^{(t)}), \quad (14)
o^(t)=σ3(mg(t)⊙ml(t)),(14)
其中
⊙
\odot
⊙表示按元素乘积。通过一个阈值
δ
\delta
δ,我们可以获得推荐的药物组合,即一个多热向量
m
^
(
t
)
\hat{m}^{(t)}
m^(t),通过选择那些值大于
δ
\delta
δ的条目。
4.4 Model Training and Inference
SafeDrug是端到端训练的。我们同时学习 E d E_d Ed、 E p E_p Ep、 R N N d RNN_d RNNd和 R N N p RNN_p RNNp中的参数、网络参数 W 1 W_1 W1、 E a E_a Ea、MPNN中的逐层参数 { W ( i ) } \{W^{(i)}\} {W(i)}、层归一化(LN)中的参数以及 W 2 W_2 W2、 W 3 W_3 W3和 W 4 W_4 W4。
多重损失在本文中,推荐任务被表述为多标签二分类。假设
∣
M
∣
|\mathcal{M}|
∣M∣是药物的总数。我们使用
m
(
t
)
m^{(t)}
m(t)表示目标多热药物推荐,
o
^
(
t
)
\hat{o}^{(t)}
o^(t)表示输出实值药物表示(与应用阈值
δ
\delta
δ后的预测多热向量
m
^
(
t
)
\hat{m}^{(t)}
m^(t)相对)。我们将每种药物的预测视为一个子问题,并使用二元交叉熵(BCE)作为损失,即:
L
b
c
e
=
−
∑
i
=
1
∣
M
∣
m
i
(
t
)
log
(
o
^
i
(
t
)
)
+
(
1
−
m
i
(
t
)
)
log
(
1
−
o
^
i
(
t
)
)
,
(
15
)
L_{bce} = -\sum_{i = 1}^{|\mathcal{M}|} m_i^{(t)} \log(\hat{o}_i^{(t)}) + (1 - m_i^{(t)}) \log(1 - \hat{o}_i^{(t)}), \quad (15)
Lbce=−i=1∑∣M∣mi(t)log(o^i(t))+(1−mi(t))log(1−o^i(t)),(15)
其中
m
i
(
t
)
m_i^{(t)}
mi(t)和
o
^
i
(
t
)
\hat{o}_i^{(t)}
o^i(t)是第
i
i
i个条目。为了使结果更稳健,我们还采用多标签铰链损失以确保真实标签比其他标签至少大1个余量,即:
L
m
u
l
t
i
=
∑
i
,
j
:
m
i
(
t
)
=
1
,
m
j
(
t
)
=
0
max
(
0
,
1
−
(
o
^
i
(
t
)
−
o
^
j
(
t
)
)
)
∣
M
∣
.
(
16
)
L_{multi} = \sum_{i,j: m_i^{(t)} = 1, m_j^{(t)} = 0} \frac{\max(0, 1 - (\hat{o}_i^{(t)} - \hat{o}_j^{(t)}))}{|\mathcal{M}|}. \quad (16)
Lmulti=i,j:mi(t)=1,mj(t)=0∑∣M∣max(0,1−(o^i(t)−o^j(t))).(16)
基于DDI邻接矩阵
D
D
D,我们还设计了关于
o
^
(
t
)
\hat{o}^{(t)}
o^(t)的不良DDI损失,即:
L
d
d
i
=
∑
i
=
1
∣
M
∣
∑
j
=
1
∣
M
∣
D
i
j
⋅
o
^
i
(
t
)
⋅
o
^
j
(
t
)
,
(
17
)
L_{ddi} = \sum_{i = 1}^{|\mathcal{M}|} \sum_{j = 1}^{|\mathcal{M}|} D_{ij} \cdot \hat{o}_i^{(t)} \cdot \hat{o}_j^{(t)}, \quad (17)
Lddi=i=1∑∣M∣j=1∑∣M∣Dij⋅o^i(t)⋅o^j(t),(17)
其中
⋅
\cdot
⋅是标量之间的乘积。请注意,上述损失函数是为单次就诊定义的。在训练期间,损失反向传播将在患者级别通过所有就诊的平均损失进行。
可控损失函数使用多个损失函数进行训练的标准方法是通过损失度量项的加权和[Dosovitskiy and Djolonga, 2019],即:
L
=
β
(
α
L
b
c
e
+
(
1
−
α
)
L
m
u
l
t
i
)
+
(
1
−
β
)
L
d
d
i
,
(
18
)
L = \beta (\alpha L_{bce} + (1 - \alpha) L_{multi}) + (1 - \beta) L_{ddi}, \quad (18)
L=β(αLbce+(1−α)Lmulti)+(1−β)Lddi,(18)
其中
α
\alpha
α和
β
\beta
β通常是预定义的超参数。
在这种情况下,两个预测损失 L b c e L_{bce} Lbce和 L m u l t i L_{multi} Lmulti是兼容的,因此我们从验证集中选择 α \alpha α。我们观察到数据集中也存在不良DDI(医生可能会错误地开具相互作用的药物)。因此,通过使用真实标签进行训练,正确的学习过程可能反而会增加DDI。因此,我们考虑通过比例 - 积分 - 微分(PID)控制器[An et al., 2018]在训练过程中调整 β \beta β,以平衡预测损失和DDI损失。为简单起见,当推荐药物的DDI率高于某些阈值时,我们仅利用比例误差信号作为负反馈。
在安全药物推荐中,关键是保持低DDI,因此我们为损失函数设置了一个DDI接受率 γ ∈ ( 0 , 1 ) \gamma \in (0, 1) γ∈(0,1)。我们的动机是,如果患者级别的DDI低于阈值 γ \gamma γ,则我们仅考虑最大化预测准确性;否则, β \beta β将自适应地调整以降低DDI。
可控因子
β
\beta
β遵循分段策略,即:
β
=
{
1
,
DDI
≤
γ
max
{
0
,
1
−
D
D
I
−
γ
K
p
}
,
DDI
>
γ
(
19
)
\beta = \begin{cases} 1, & \text{DDI} \leq \gamma \\ \max \{0, 1 - \frac{DDI - \gamma}{K_p}\}, & \text{DDI} > \gamma \end{cases} \quad (19)
β={1,max{0,1−KpDDI−γ},DDI≤γDDI>γ(19)
其中
K
p
K_p
Kp是比例信号的校正因子。
我们在实验中表明, γ \gamma γ可以被视为输出DDI率的上限。通过预设 γ \gamma γ,我们的SafeDrug可以满足不同水平的DDI要求。
模型推理阶段基本上遵循与训练相同的流程。我们在方程(14)中的输出药物表示上应用阈值 δ = 0.5 \delta = 0.5 δ=0.5,并选择对应于值大于 δ \delta δ的条目的药物作为最终推荐。我们在附录中构建了整体算法。
5 Experiments
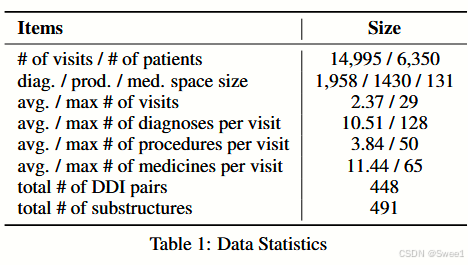
数据集和指标实验在MIMIC - III[Johnson等人,2016]上进行。后处理数据的统计信息在表1中报告。我们使用五个有效性指标:DDI率、Jaccard相似度、F1分数、PRAUC和药物数量,来评估推荐效率;以及三个模型复杂度指标:参数数量、训练时间和推理时间。由于篇幅限制,其他细节推至附录。
基线我们从不同角度将SafeDrug与以下基线进行比较:标准逻辑回归(LR)、多标签分类方法:集成分类链(ECC)、[Read等人,2009]、基于RNN的方法:RETAIN[Choi等人,2016b]、基于实例的方法:LEAP[Zhang等人,2017]、基于纵向记忆的方法:DMNC[Le等人,2018]和GAMENet[Shang等人,2019]。由于[Shang等人,2018]需要额外的本体数据,因此不将其视为基线。我们还将SafeDrug与其两个模型变体进行了比较:
S
a
f
e
D
r
u
g
L
SafeDrug_L
SafeDrugL是仅具有二分编码器(局部,L)的模型,
S
a
f
e
D
r
u
g
G
SafeDrug_G
SafeDrugG是仅具有MPNN编码器(全局,G)的模型。在实现中,我们分别使用
m
l
(
t
)
m_l^{(t)}
ml(t)和
m
g
(
t
)
m_g^{(t)}
mg(t)作为它们的最终药物表示。
性能比较从表2可以很容易看出,药物序列生成模型LEAP、DMNC和ECC在基线中效果不佳。因此,我们得出结论,将药物推荐任务表述为多标签预测可能更直接有效。LR和RETAIN没有考虑DDI信息,所以它们输出的DDI率不理想。GAMENet给出的推荐是基于历史组合,然而,实际的药物记录通常包含较高的DDI率,例如在MIMIC - III中约为8%,所以它也提供了较高的DDI率。此外,两个变体
S
a
f
e
D
r
u
g
L
SafeDrug_L
SafeDrugL和
S
a
f
e
D
r
u
g
G
SafeDrug_G
SafeDrugG的性能表明,两种分子编码器都有助于预测。总体而言,我们的SafeDrug在DDI率显著更低且准确性更好的情况下,也优于其他基线。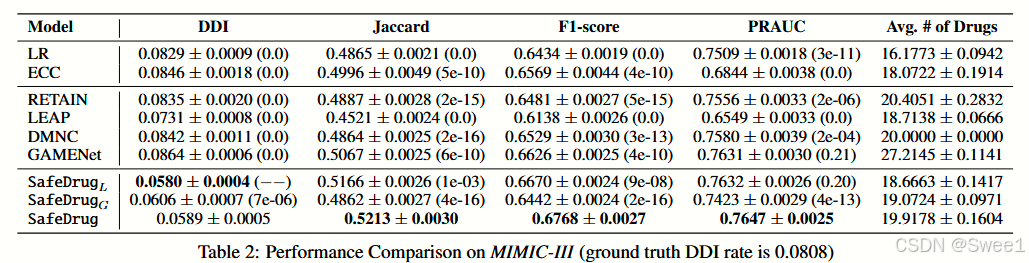
显著性检验上述结果是通过10轮自助抽样给出的。接下来,我们对每个两尾假设的指标进行T检验。
p
p
p值显示在括号中。通常,小于0.05的
p
p
p值将被视为显著。对于SafeDrug的结果比基线差的情况,我们也使用(–)表示。总之,我们的模型在几乎每个指标上都能以
p
=
0.001
p = 0.001
p=0.001的显著水平击败所有比较的基线。
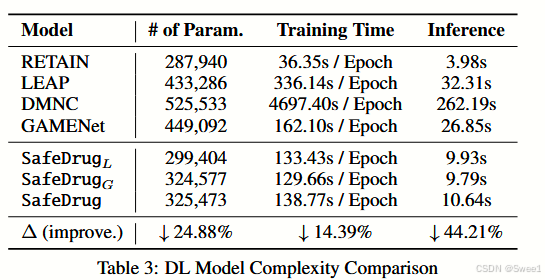
模型效率比较SafeDrug和其他深度学习基线的模型复杂度在表3中进行评估。由于RETAIN并非专门用于药物推荐,表3中的改进是相对于LEAP、DMNC和GAMENet而言的。在效率方面,我们的模型相对于LEAP、DMNC和GAMENet具有相对较低的空间和时间复杂度。而且,它在推理过程中效率更高。LEAP和DMNC采用顺序建模,逐个推荐药物,这很耗时。GAMENet存储了一个大型记忆库,因此需要更大的空间。相比之下,我们得出结论,SafeDrug高效且灵活,可用于实际部署。
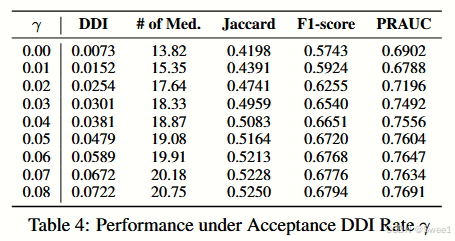
DDI可控性分析我们评估了DDI可控性,并表明我们输出的平均DDI率可以通过阈值
γ
\gamma
γ进行控制。MIMIC - III中的真实DDI率为0.0808。我们认为基于学习的药物推荐方法应该通过处理电子健康记录(EHR)和药物 - 药物关系数据库来优化最终推荐,因此输出DDI率应该低于人类专家的水平。在本节中,我们针对一系列目标DDI
γ
\gamma
γ(范围从0到0.08)测试了一个模型。对于每个
γ
\gamma
γ,我们训练一个单独的模型。实验进行了5次。收敛后,我们在表4中展示了平均指标。
从结果来看,DDI率受到
γ
\gamma
γ的控制且有上限,这在大多数情况下与设计一致。当
γ
\gamma
γ变大时,一个组合中允许的药物更多,SafeDrug的准确性更高。当
γ
\gamma
γ太小(<
0.02
0.02
0.02)时,模型准确性会急剧下降。参数
γ
\gamma
γ为医生提供了一种在推荐中控制DDI率和准确性之间权衡的方法。
6 Conclusion
在本文中,我们提出了SafeDrug,用于准确且安全的药物推荐。我们的模型提取并编码丰富的分子结构信息,以增强药物推荐任务。我们设计了一种自适应损失控制器,以提供额外的灵活性。我们使用基准数据对SafeDrug进行了评估,结果显示其具有更好的准确性和效率。
致谢
这项工作部分得到了美国国家科学基金会奖项SCH - 2014438、PPoSS 2028839、IIS - 1838042,美国国立卫生研究院奖项NIH R01 1R01NS107291 - 01以及OSF Healthcare的支持。



















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











