









虽然说R有许多GEO数据挖掘的包和库,但是我不怎么喜欢R语言,此时,Python的rpy2库就派上用场了!
目录
1.构建分组信息
以GSE5281为例,在下载数据时,还有一个临床信息的数据,从中可以知道,哪些是AD,哪些是Control
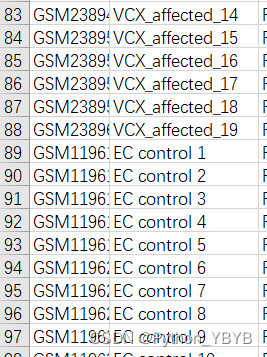
该数据从1—87是AD样本,88—161是Control样本,我们可以自建分组信息:
导入需要用到的库
import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
import rpy2
from rpy2.robjects import r
from rpy2.robjects.packages import importr
from rpy2.robjects import pandas2ri
pandas2ri.activate导入数据
df=pd.read_excel("F:\\bioinformatics\\20220806\\data\\GSE5281\\NEW_GSE5281.xlsx")
#将symbol设置为索引列
df.set_index('symbol',inplace=True)
print(df.head())
自建分组信息
##提取列索引
list1=df.columns.tolist()
#修改一下样本ID的符号
list11=[]
for i in list1:
a=i+"_log"
list11.append(a)建一个分组信息的列表与上述列表组建一个dataframe数据框
list2=['AD']*87+['Control']*74
data={'sample_id':list11,'group_list':list2}
group_inf=pd.DataFrame(data)
print(group_inf)这样就完成了分组信息的构建

2.检查表达矩阵
(1)检查数据是否经过了log处理以及归一化化
为什么要对表达矩进行log对数转换?见博客文章:
https://blog.csdn.net/tuanzide5233/article/details/88542805
GEO芯片数据差异表达分析时是否需要log2以及标准化的问题
https://blog.csdn.net/tuanzide5233/article/details/88542558
抽取一些基因看看表达量
print(df.loc[['GAPDH','ACTB','ZYG11B']])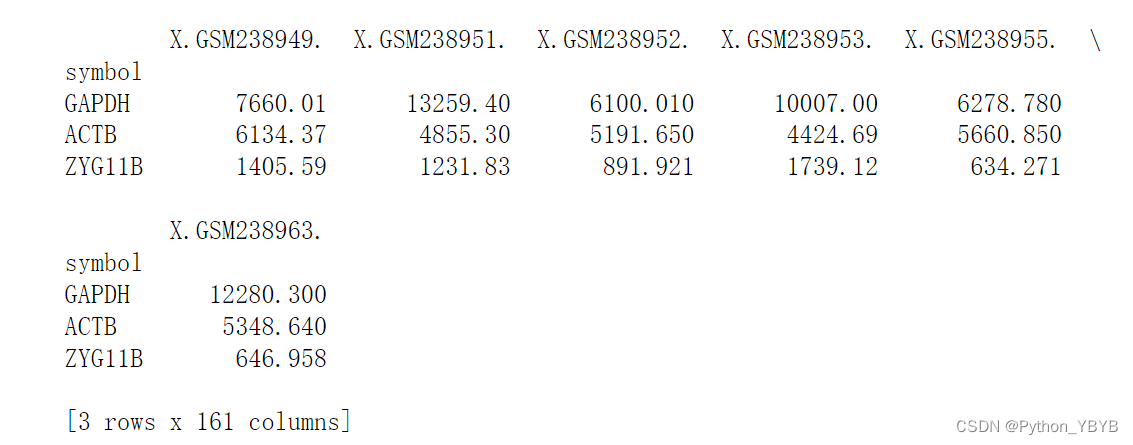
都已经达到五、六千甚至上万了,肯定没有经过取对数处理,因为2的6000次方有多大难以想象!
那就对表达矩阵取log2:
columns=df.columns
for i in columns:
df[i+"_log"]=df[i].map(lambda x : np.log2(x)) #做对数转换
df1=df.loc[:,df.columns.str.contains('log')] #切片转换数据
print(df1.head())
这样就经过了对数log2转换。
接下画箱形图看看数据是否通过了归一化
plt.figure(figsize=(10,5)) #设置画布尺寸
plt.title('Example of boxplot',fontsize=20) #设置标题,并设置字号大小
df1.boxplot()
plt.show()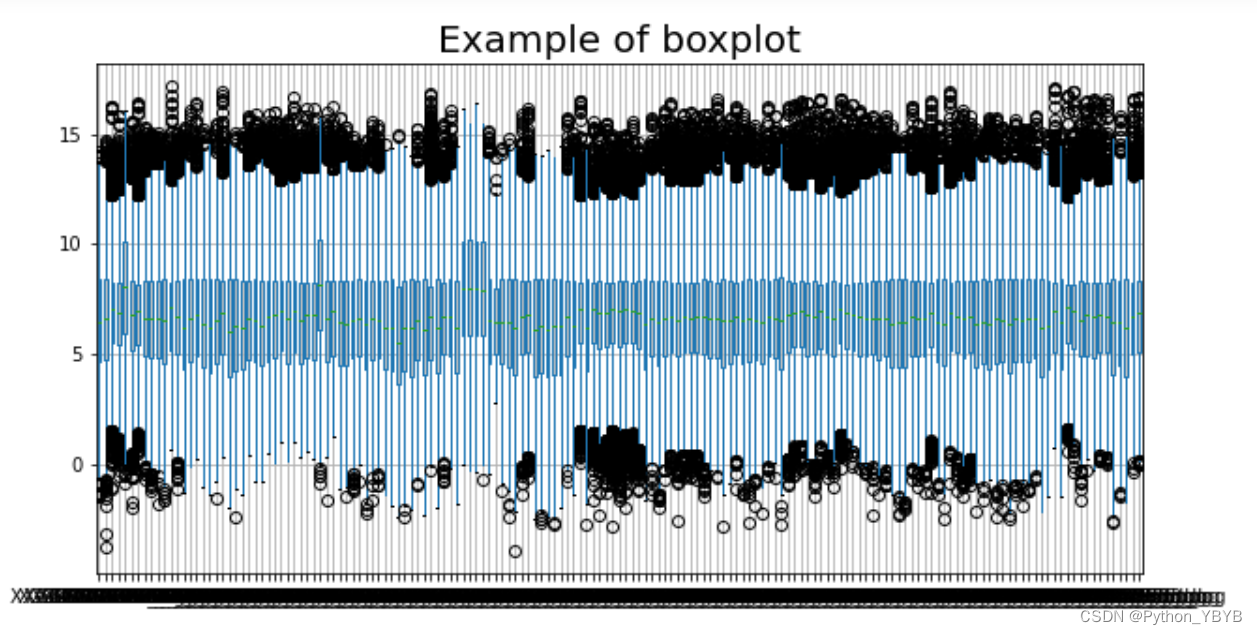
注意!这里不能用df1.iloc[[1,2,3],[5,6,7,8,9]]去看,因为不能体现数据的整体性。从图中可以看出,数据并一致性并不好。可以用rpy2对接R语言采用limma包中normalizeBetweenArrays函数来标准化数据 :
limma=importr("limma") ##加载R的limma包
normalized_exp=limma.normalizeBetweenArrays(df1) #做分位数标准化处理
normalized_exp上述代码等价于
from rpy2.robjects import pandas2ri
pandas2ri.activate()
rdf=pandas2ri.py2rpy(df1)
from rpy2.robjects import globalenv ##让globalenv来激活rdf
importr("limma")
globalenv['rdf']=rdf
rscript="""
normalized_exp=normalizeBetweenArrays(rdf)
"""
result=r(rscript)
result
发现返回的是narray矩阵,加一行代码还原成dataframe数据框
df2=pd.DataFrame(data=normalized_exp[0:,0:],index=df.index,columns=list11) #还原成原来的dataframe数据框
print(df2.head())
再画箱线图看看数据分布情况
from jupyterthemes import jtplot ##使得图形适应背景
jtplot.style()
plt.figure(figsize=(10,5)) #设置画布尺寸
plt.title('Example of boxplot(normalized)',fontsize=20) #设置标题,并设置字号大小
df2.boxplot()
plt.show()
发现大部分基因的表达量在6左右
(2)检验常见基因的表达量(如:GAPDH、ACTB)
查看一些管家基因的表达量,如果表达高于正常值,就说明数据没有问题
print(df2.loc[['GAPDH','ACTB']])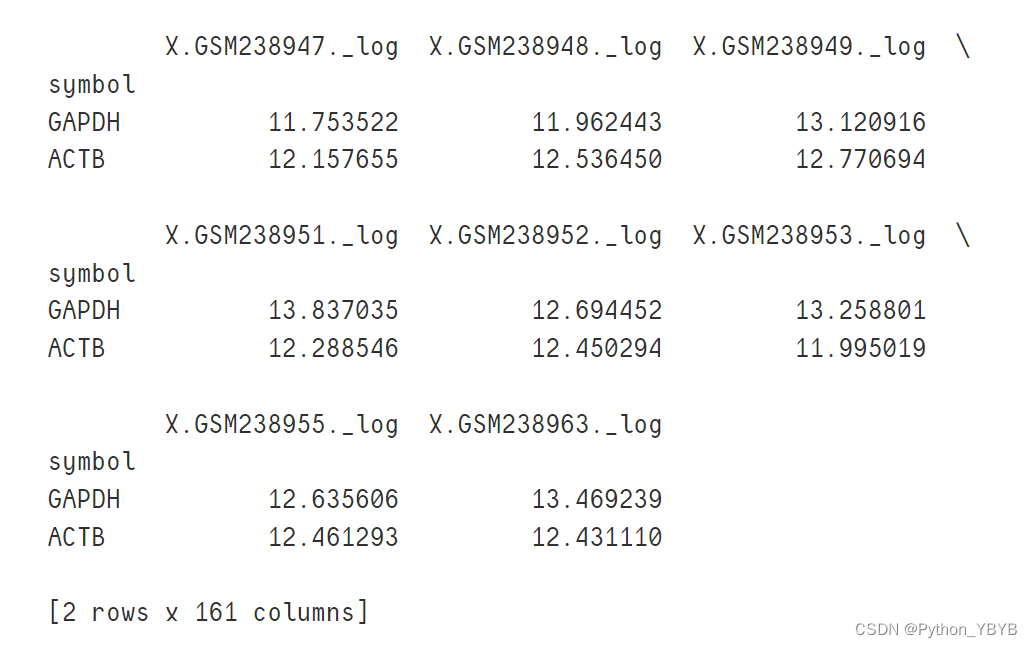
一看大多数在12左右,表达量比大多数基因高,说明符合预期。假如发现管家基因的表达量特别低,那就要考虑是不是之前的代码出了问题,比如ID转换..............
(3)检查样本分组信息
检查样本分组信息,一般看PCA降维,hclust图
先看凝聚层次聚类
import scipy.cluster.hierarchy as shc
plt.figure(figsize=(10,7)) #设置画布尺寸
plt.title("sample hcluster")
dend=shc.dendrogram(shc.linkage(df2.T,method='ward'),labels=df2.columns)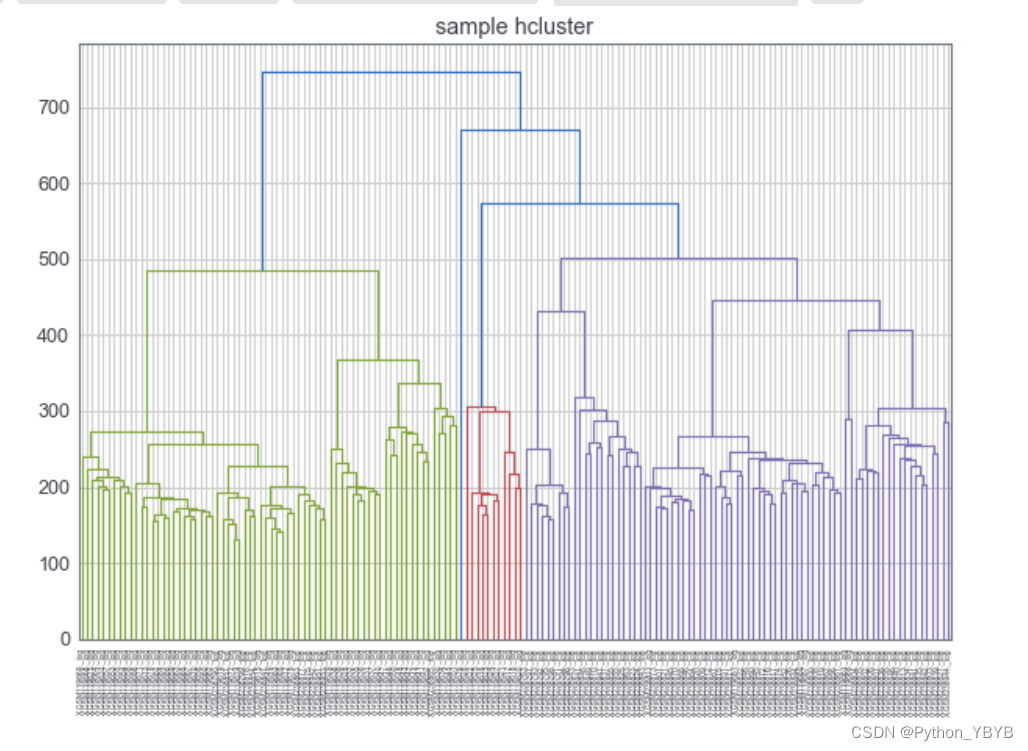
图形出来后发现样本比较大看不出来,可以作随机抽样看看
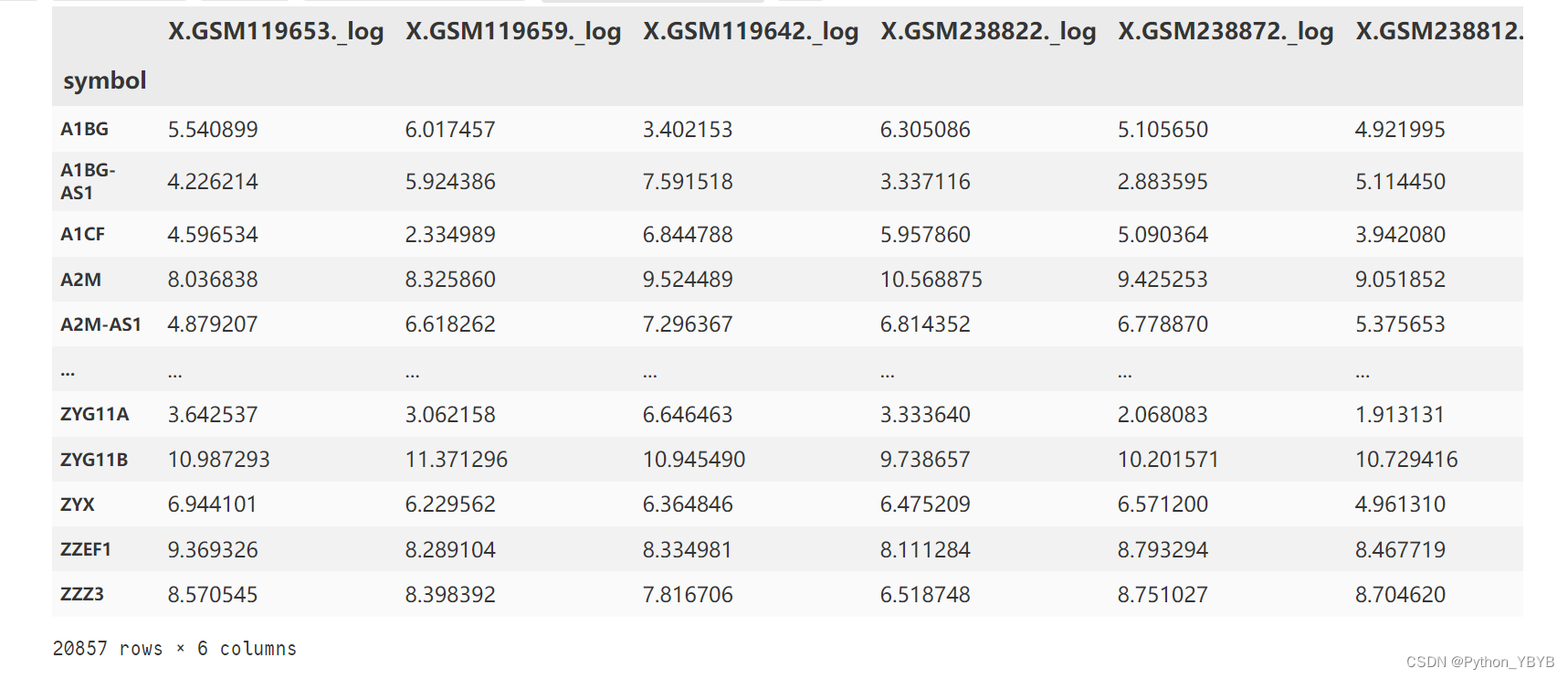
d1=df2.iloc[0:,:87] #抽取AD数据
d2=df2.iloc[0:,87:162] #抽取Control数据
##在两组中作随机抽样
d11=d1.sample(n=3,random_state=1,replace=False,axis=1)
d22=d2.sample(n=3,random_state=1,replace=False,axis=1)
d=pd.merge(d11,d22,on='symbol') #合并抽样所得到的数据
d抽样得到的数据
画出来层次聚类数看看
plt.figure(figsize=(10,7)) #设置画布尺寸
plt.title("sample hcluster2")
dend=shc.dendrogram(shc.linkage(d.T,method='complete'),labels=d.columns)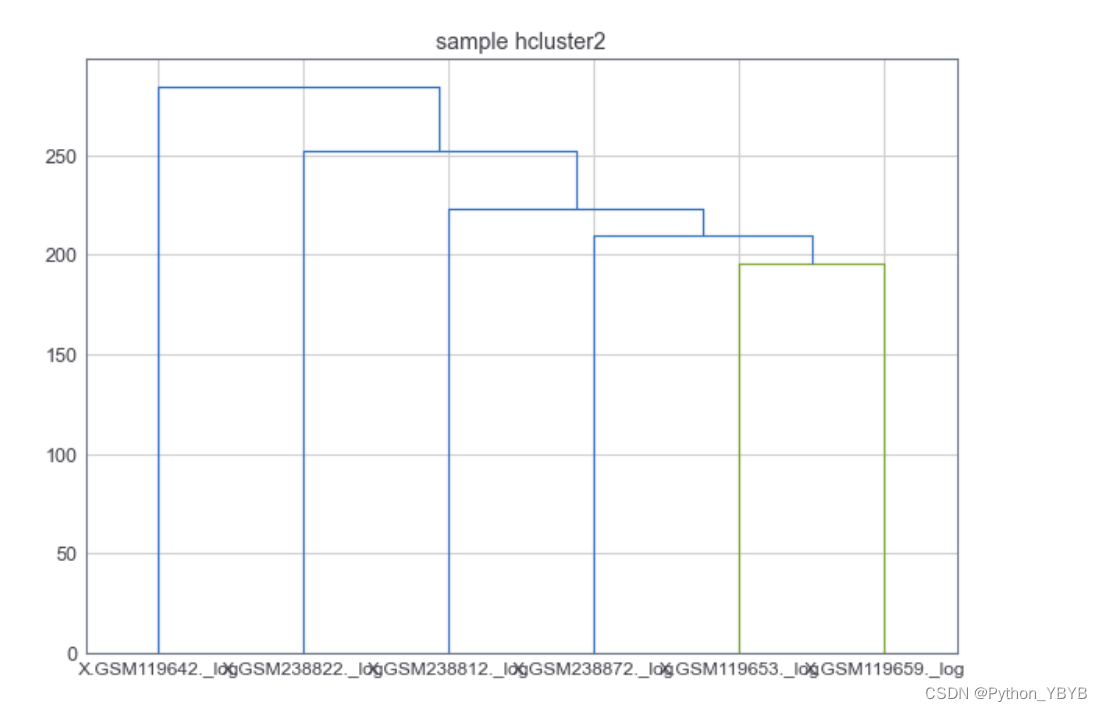
感觉不怎么分得开,再来以下PCA瞧瞧:
from sklearn.decomposition import PCA
pca=PCA(n_components=2) ##设置维度为2
reduced_X=pca.fit_transform(df2.T)
plt.scatter([x[0] for x in reduced_X[0:86]],[x[1] for x in reduced_X[0:86]],c="r")
plt.scatter([x[0] for x in reduced_X[87:162]],[x[1] for x in reduced_X[87:162]],c="g")
plt.xlabel('PC1')
plt.ylabel('PC2')
plt.legend(['AD','Control'])
plt.title('PCA')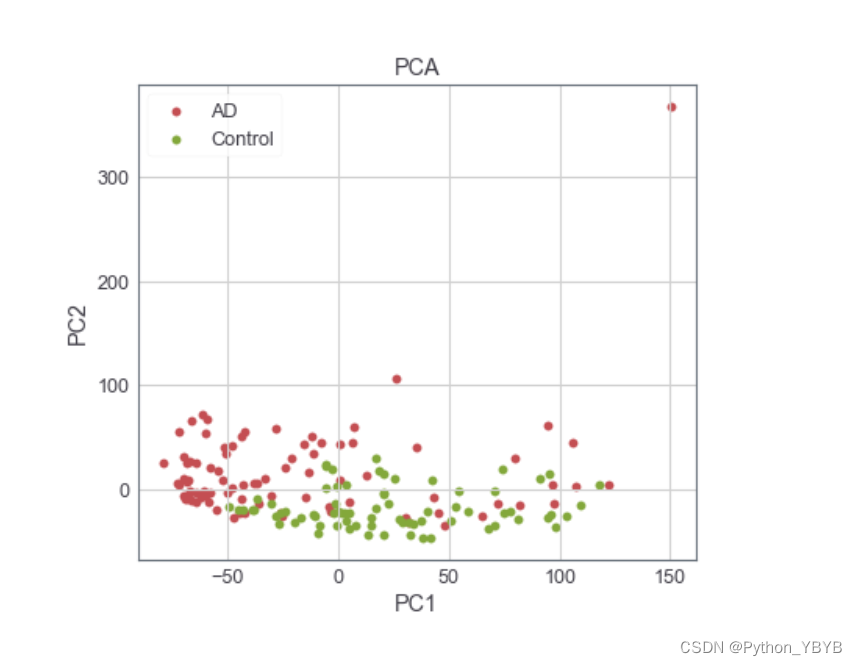
同样分的也不是很好,也没有办法,就这个样子了,望有好的解决办法的大神指教!这部分内容就到这里。



















 1840
1840

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











