library(tidyverse)
qpcr_count <- function(raw_data,ref_sample,ref_gene) {
colnames(raw_data) = c("Sample Name","Target Name","CT" )
raw_data$CT = raw_data$CT %>% as.numeric()
Treat_samples = raw_data$`Sample Name` %>% unique() %>% setdiff(ref_sample)
genes = raw_data$`Target Name` %>% unique() %>% setdiff(ref_gene)
result = raw_data %>%left_join(
raw_data %>%
filter(`Target Name` == ref_gene) %>%
group_by(`Sample Name`) %>%
mutate(mean_ref_gene = mean(CT)) %>%
dplyr::select(`Sample Name`,mean_ref_gene) %>%
unique(),
by =c("Sample Name")
) %>%
mutate(DeltaCT = CT - mean_ref_gene) %>%
left_join(
raw_data %>%left_join(
raw_data %>%
filter(`Target Name` == ref_gene) %>%
group_by(`Sample Name`) %>%
mutate(mean_ref_gene = mean(CT)) %>%
dplyr::select(`Sample Name`,mean_ref_gene) %>%
unique(),
by =c("Sample Name")
) %>%
mutate(DeltaCT = CT - mean_ref_gene) %>%
filter(`Sample Name` == ref_sample) %>% group_by(`Target Name`) %>%
mutate(CK_sample_mean_DeltaCT = mean(DeltaCT)) %>% ## 分别计算对照组每个基因的DeltaCT的均值
dplyr::select(`Target Name`,CK_sample_mean_DeltaCT) %>%
unique(),by =c("Target Name")
) %>%
mutate(DeltaDeltaCT = DeltaCT - CK_sample_mean_DeltaCT) %>% ## 计算DeltaDeltaCT
mutate(RelativeExpression = 2^(-DeltaDeltaCT)) %>% ## 计算2^(-DeltaDeltaCT)
dplyr::select("Sample Name","Target Name","CT","DeltaCT" ,"DeltaDeltaCT","RelativeExpression" )
return(result)
}
library(readxl)
qocr <- read_excel("~/Program/wanggang/螺/qocr.xlsx")
qocr$`Sample Name` = qocr$`Sample Name` %>% str_sub(1,1)
qpcr_data =qocr
qpcr_res = qpcr_data %>%qpcr_count(ref_sample = "L",ref_gene = "ACTIN")
qpcr_res_sum =qpcr_data %>%
qpcr_count(ref_sample = "L",ref_gene = "ACTIN") %>%
filter(`Sample Name` == "L") %>%
group_by(`Target Name`) %>%
summarise(mean = mean(RelativeExpression),
sd = sd(RelativeExpression)) %>%
mutate(`Sample Name` = "L") %>%
bind_rows(
qpcr_data %>%
qpcr_count(ref_sample = "L",ref_gene = "ACTIN")%>%
filter(`Sample Name` != "L") %>%
group_by(`Target Name`) %>%
summarise(mean = mean(RelativeExpression),
sd = sd(RelativeExpression)) %>%
mutate(`Sample Name` = "J")
)
qpcr_res_sum$`Sample Name` = qpcr_res_sum$`Sample Name` %>% factor(levels = c("L","J"))
qpcr_res$`Sample Name`= qpcr_res$`Sample Name` %>% factor(levels = c("L","J"))
c("ACTIN","SOX2","SOX10","WNT2","WNT7","FOXL2")
qpcr_res_sum$`Sample Name` = qpcr_res_sum$`Sample Name` %>% str_replace_all("L","ovaries") %>% str_replace_all("J","testes")
qpcr_res$`Sample Name` = qpcr_res$`Sample Name` %>% str_replace_all("J","testes")%>% str_replace_all("L","ovaries")
genenames = qpcr_res_sum$`Target Name` %>% unique() %>% setdiff("ACTIN")
qpcr_res_sum1 = qpcr_res_sum %>% filter(`Target Name` %in% c("ACTIN",genenames[1:5]) )
qpcr_res1 = qpcr_res %>% filter(`Target Name` %in% c("ACTIN",genenames[1:5]) )
library(ggsignif)
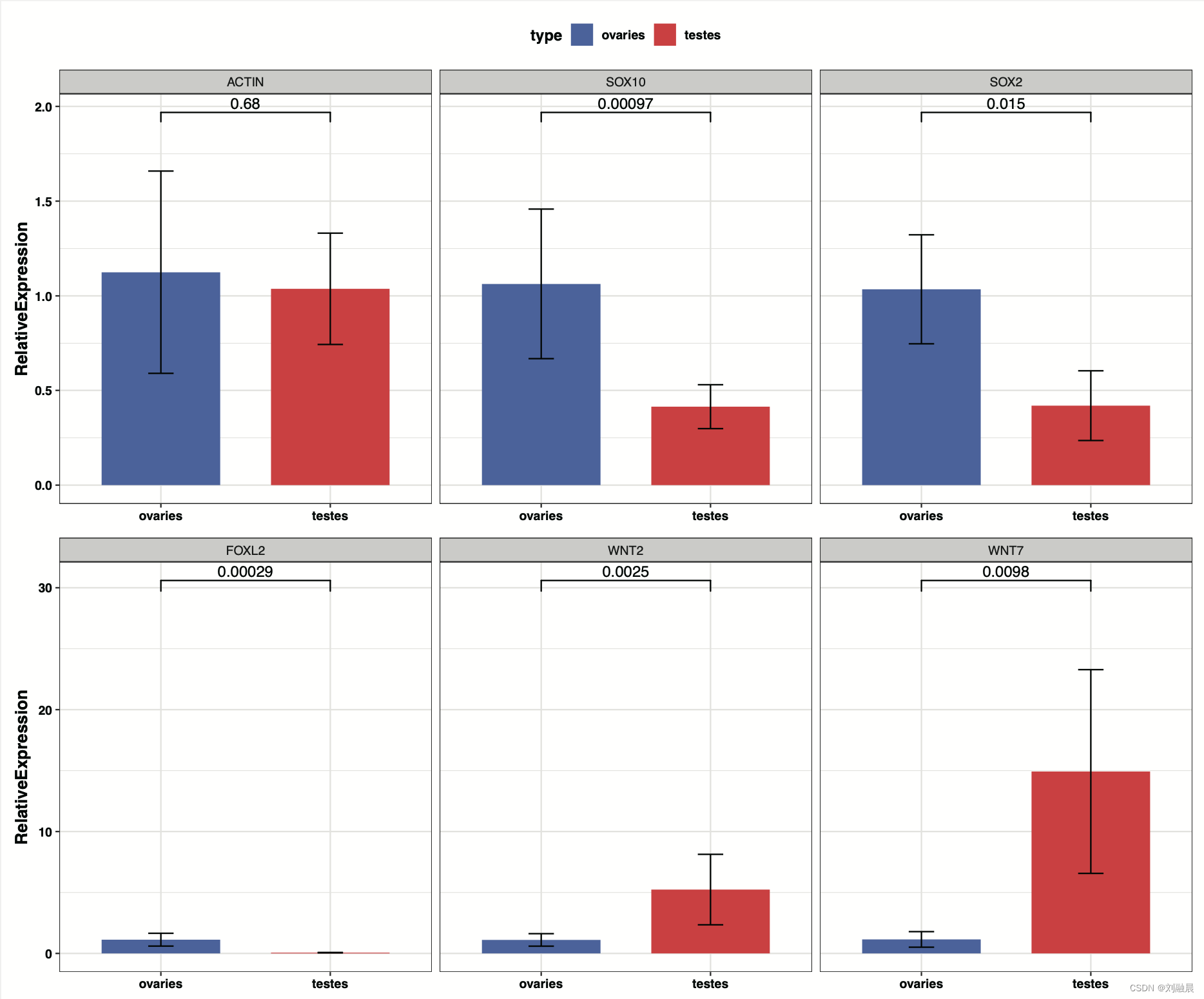
p1 = ggplot(data = qpcr_res_sum1 ,
aes(x = `Sample Name` , y = mean))+
geom_col(
aes(x = `Sample Name` , y = mean,fill = `Sample Name`),
width = 0.7)+
geom_errorbar(aes(ymin = mean - sd,
ymax = mean + sd),
fill = "black",
width = 0.15)+
geom_signif(data = qpcr_res1,
aes(x = `Sample Name`,
y = RelativeExpression),
comparisons = list(c("ovaries", "testes")),
test = "t.test",
map_signif_level = F #是否显示*号代表差异性
)+
scale_fill_manual(values = c('royalblue', '#FF0124')) +
theme_bw()+
facet_grid(. ~ `Target Name` ) +
theme(legend.position = "none") +
labs(x = NULL, y = "RelativeExpression") +
theme(axis.title = element_text(size = 12, face = "bold"), axis.text = element_text(face = "bold",colour = "gray0"),
plot.title = element_text(size = 15)) + theme(plot.title = element_text(face = "bold"),
legend.text = element_text(face = "bold"),
legend.title = element_text(face = "bold"),
legend.position = "top", legend.direction = "horizontal") +
labs(fill = "type")



























 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











