






 本文介绍了如何使用R语言中的WGCNA库进行基因表达数据的模块聚类分析,包括计算相关系数、邻接矩阵、TOM矩阵,以及模块的聚类、合并和特征向量计算。最后展示了结果,包括模块数量、大小和与样本属性的相关性。
本文介绍了如何使用R语言中的WGCNA库进行基因表达数据的模块聚类分析,包括计算相关系数、邻接矩阵、TOM矩阵,以及模块的聚类、合并和特征向量计算。最后展示了结果,包括模块数量、大小和与样本属性的相关性。
library(WGCNA)
library(tidyverse)
result = data.frame()
net_list = list()
enableWGCNAThreads(nThreads = 16) #电脑可以多线程运行
for (m in 1:4) {
Split = m
for (n in c(20,40,80,100)) {
Minnum = n
for ( i in c(0.1,0.25,0.3)) {
CutHeight = i
net <- blockwiseModules(
# 0.输入数据(表达矩阵)
datExpr,
# 1. 计算相关系数 (基因与基因之间的数据,是连续型选p)
corType = "pearson", # 相关系数算法,pearson|bicor
# 2. 计算邻接矩阵
power = 6, # 前面得到的 soft power
networkType = "unsigned", # unsigned | signed | signed hybrid
# 3. 计算 TOM 矩阵 (生成TOM的距离矩阵)
TOMType = "unsigned", # none | unsigned | signed
saveTOMs = TRUE,
saveTOMFileBase = "blockwiseTOM",
# 4. 聚类并划分模块
deepSplit = Split, # 0|1|2|3|4, 值越大得到的模块就越多越小
minModuleSize = Minnum, #模块里面最少有多少基因
# 5. 合并相似模块
## 5.1 计算模块特征向量(module eigengenes, MEs),即 PC1
## 5.2 计算 MEs 与 datTrait 之间的相关性
## 5.3 对距离小于 mergeCutHeight (1-cor)的模块进行合并
mergeCutHeight = CutHeight, #模块与模块之间相关性>0.75将两个模块合并
# 其他参数
numericLabels = FALSE, # 是否以数字命名模块
nThreads = 0, # 0 则使用所有可用线程
maxBlockSize = 100000 # 需要大于基因的数目
)
name = str_c(m,n,i,sep = "_")
net_list[[name]] = net
wgcna_result <- data.frame(gene_id = names(net$colors),
module = net$colors)
moduleTraitCor <- cor(
net$MEs, ##ME指模块特征向量
datTraits$sex %>% as.numeric(), #样本信息表
use = "p",
method = 'spearman' # 注意相关系数计算方式
)
moduleTraitCor = moduleTraitCor %>%
as.data.frame() %>%
rownames_to_column(var = "module")
moduleTraitCor <- moduleTraitCor[order(abs(moduleTraitCor$V1), decreasing = TRUE), ]
my_modules <- c(moduleTraitCor[1,1] %>% str_replace_all("ME",""))
# 提取该模块的表达矩阵
m_wgcna_result <- filter(wgcna_result, module %in% my_modules)
m_datExpr <- datExpr[, m_wgcna_result$gene_id]
m_datExpr = m_datExpr %>% as.data.frame() %>% t()%>%
as.data.frame() %>% rownames_to_column(var = "gene_id")
cor = moduleTraitCor$V1[1]
res = data.frame(deepSplit = Split,
minModuleSize = Minnum,
mergeCutHeight = CutHeight,
module_num = m_datExpr %>% nrow(),
cor = cor)
res$deepSplit = res$deepSplit %>% as.character()
res$minModuleSize = res$minModuleSize %>% as.character()
res$mergeCutHeight = res$mergeCutHeight %>% as.character()
res$module_num = res$module_num %>% as.character()
res$cor =res$cor %>% as.character()
result =result %>% bind_rows(res)
}
}
}
save(net_list,file = "./wgcna.Rdata")
write_csv("./wgcna_result.csv")最终结果如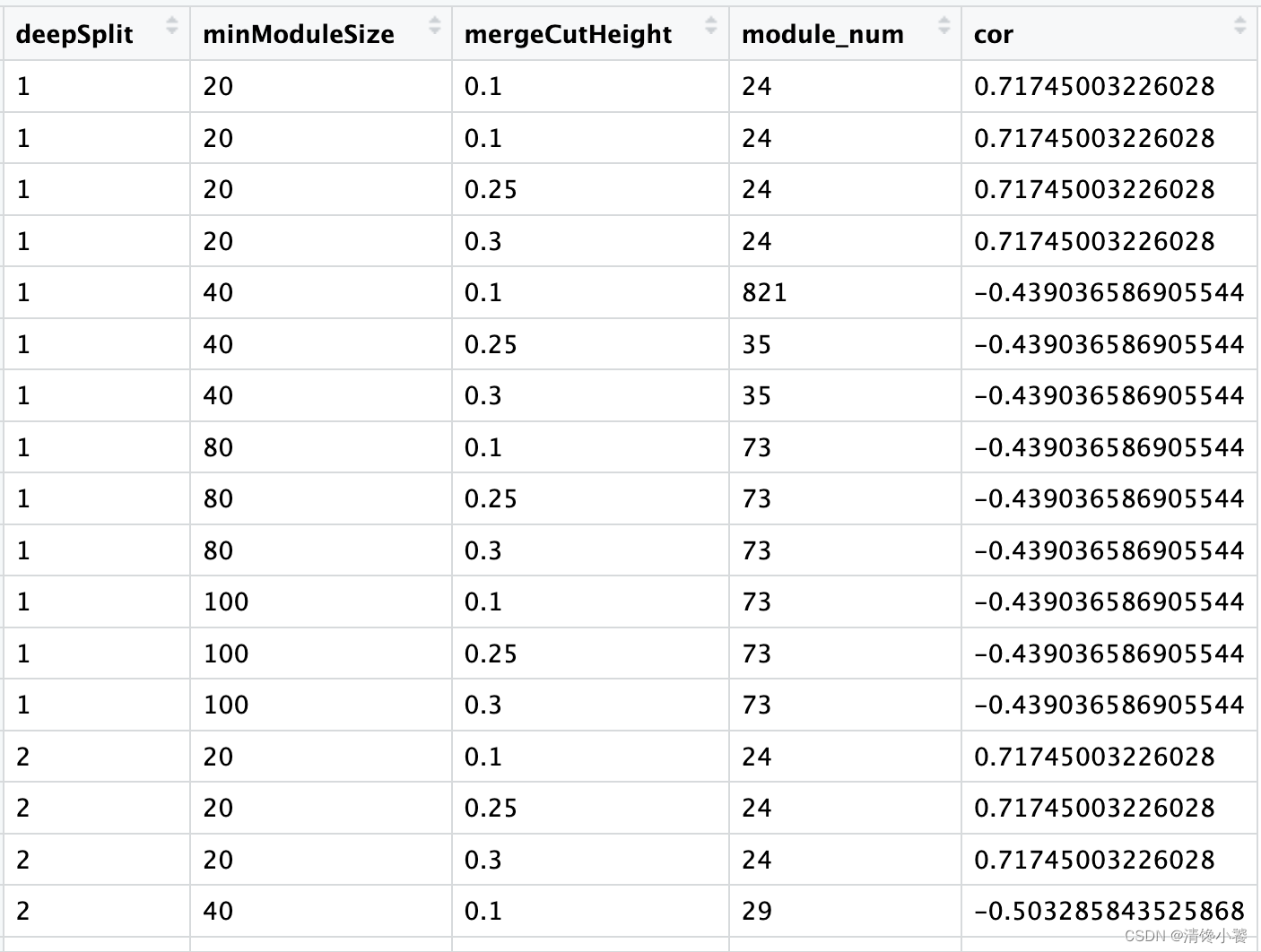
















 699
699











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











