



监管机构及其在医疗器械和伤口敷料方面的作用
15.1 引言
医疗器械在医疗保健行业中的重要性日益增加。目前,市场上有超过8000种通用医疗器械,其中一些器械含有药物[1],这些需求要求各国持续审查和监管监管框架。这确保了产品能够实现预期的效益,并且对人体使用是安全的。
对于开发和生产医疗器械的企业而言,一个主要问题是当前的监管需求必须根据法规要求进行更新,并在生产过程中予以实施[2]。全球医疗器械市场的价值估计为2280亿美元,预计从2010年的1640亿美元增长到2018年的约4400亿美元。其复合年增长率约为每年4.4%。美国是全球最大的医疗器械市场之一,其价值约为1254亿美元,占全球医疗器械市场的38%2012[3]。欧洲联盟(欧盟)占据全球第二大医疗器械市场,估计价值为1000亿欧元。主要推动国家包括德国、法国、意大利、英国和西班牙[4]。中国是全球第三大医疗器械市场,自2009年以来年均增长率达20%,2015年估值超过480亿美元2012[5]。全球医疗器械市场份额如图151[6]所示。
表15.1列出了全球领先的前20家医疗器械制造公司。美敦力公司是全球领先的医疗器械公司,2016年收入达288亿美元,位居第一;强生以251亿美元的销售额位居第二。通用电气医疗集团以183亿美元的市场份额排名第三,西门子医疗以152亿美元的市场份额紧随其后。[7]。
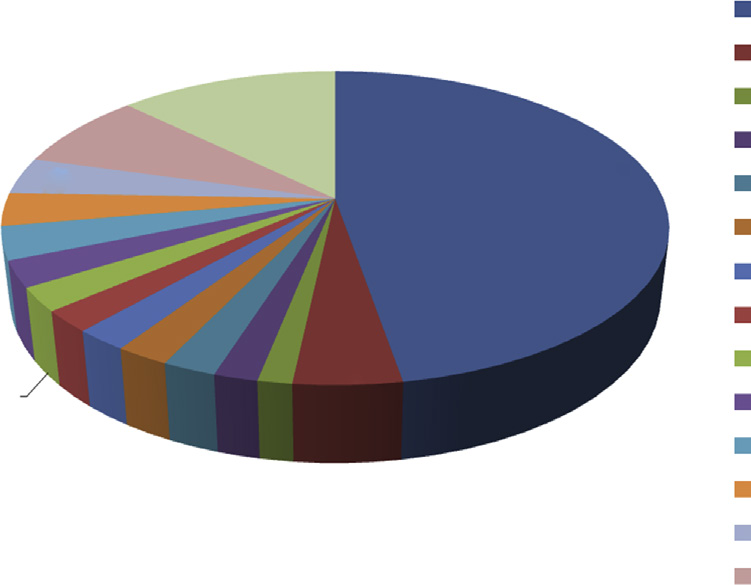
表15.1 基于2016财年销售报告的全球前20大医疗器械制造公司
| Rank | 公司 | 价值(十亿美元) |
|---|---|---|
| 1. | 美敦力公司 | 28.8 |
| 2. | 强生 | 25.1 |
| 3. | 通用电气医疗 | 18.3 |
| 4. | 西门子医疗 | 15.2 |
| 5. | 贝克顿‐迪金森 | 12.5 |
| 6. | 康德乐健康 | 12.4 |
| 7. | 飞利浦健康科技 | 12.4 |
| 8. | 史赛克 | 11.3 |
| 9. | 百特 | 10.2 |
| 10. | 雅培实验室 | 10.1 |
| 11. | 波士顿科学 | 8.4 |
| 12. | 丹纳赫 | 7.8 |
| 13. | 齐默尔毕奥美特 | 7.7 |
| 14. | 依视路 | 7.5 |
| 15. | B·布朗 | 6.8 |
| 16. | 圣犹达医疗 | 6.0 |
| 17. | 爱尔康 | 5.8 |
| 18. | 3M 医疗 | 5.5 |
| 19. | 费森尤斯 | 5.4 |
| 20. | 欧林巴斯 | 5.4 |
15.2 医疗器械
医疗器械是指任何单独使用或组合使用的仪器、装置、用具、材料或其他物品,包括制造商规定为正常使用所必需的软件。医疗器械是一种用于患者医疗目的(诊断,
医疗器械市场份额
12.7
2.7 2.4 2.2 2.2 2.2 2.2 1.8 1.4 4.4
7.9 3.8
33..44
47.3
其他医疗器械 医疗器械零部件及配件 牙科修复体 助听器 计算机断层扫描设备 眼科设备和仪器 机械疗法和心理能力测试 医用家具 牙科设备和器械 起搏器 注射器和针头 治疗性呼吸设备 X射线设备 骨科和骨折设备
治疗或手术)。当应用于人体时,医疗器械的作用主要是物理性的,与药品的生化效应相反。医疗器械在人体内或人体上的主要预期作用不是通过药理学、免疫学或代谢方式进行的,但其功能可借助这些方式辅助实现[8]。此定义包括用于给予药品的器械,例如注射泵,或包含被定义为药品的物质的器械,例如药物洗脱支架。
世界卫生组织将医疗器械定义为
任何仪器、装置、器具、机器、用具、植入物、体外诊断试剂、软件、材料或其他类似或相关物品,由制造商预期单独或组合使用于人体,用于以下一种或多种特定医疗用途:疾病的诊断、预防、监控、治疗或缓解;损伤的诊断、监控、治疗、缓解或补偿;解剖结构或生理过程的检查、替代、调节或支持;维持生命;避孕控制;医疗器械消毒;通过体外检查人体样本提供信息;且其主要预期作用不是通过药理学、免疫学或代谢方式在人体内或体表实现,但其预期功能可借助此类方式辅助实现[9]。
医疗器械的示例包括从普通伤口敷料到生命支持设备:
- 血压监测仪、血糖仪、体温计、电极、外部假体、固定胶带、加压绷带、伤口敷料、隐形眼镜、导尿管、阴道内和肠内装置[胃管、乙状结肠镜(检查S形结肠)、结肠镜(检查结肠)、胃镜(检查胃)],气管插管(检查气管)、支气管镜(检查支气管)、牙科修复体、正畸装置、宫内节育器、溃疡、烧伤和肉芽组织敷料或愈合装置以及闭塞性贴片;
- 测量神经和肌肉产生的电场强度的器械,如心电图仪(ECG)、生物电抗——常用于婴儿呼吸暂停(呼吸停止)监控,腹腔镜、关节镜(检查关节内部,如膝关节)、引流系统、牙科水泥、牙科填充材料和皮肤缝钉;
- 心脏起搏器、除颤器、心脏瓣膜、血管移植物、体内药物输送导管、心室辅助装置、神经肌肉传感器和模拟器、替代肌腱、乳房植入物、人工喉(咽部与气管之间的呼吸道)、体外氧合器管路及附件、骨科销钉、钢板、关节替代物、骨假体、骨内装置(通过骨皮质穿刺进入骨髓腔,以立即接入循环系统);
- 失禁产品(例如成人纸尿裤)、用于治疗乳头内陷的吸奶器、具有医疗宣称的肌肉紧致产品(如治疗失禁)、用于治疗临床肥胖的瘦身产品以及宣称具有止痛功效的外用热敷垫;
- 辅助技术产品,如轮椅、助行架/站立架、拐杖、视障人士移动辅助器具、康复三轮车/代步车、骨科鞋、肢体外部假肢及其附件以及矫形器(下肢/上肢、脊柱、腹部、颈部、头部)。
- 运动支撑产品:冷热敷垫、扭伤绷带、加压绷带、投放市场的健身器材,特别是用于测量心率或呼吸频率的器械——血压监测仪即使 intended 在健身房使用,也会被视为医疗器械;
- 软件,例如旨在增强X射线或超声波图像的软件,将被视为医疗器械。而仅用于患者管理或记录存储的软件则不会被视为医疗器械;以及
- 成为医疗器械附件的产品:示例包括与医疗设备配合使用的灭菌器、用于包装再灭菌医疗器械的袋、电池驱动的电子医疗器械专用的电池充电器、隐形眼镜护理产品、特定用途的医疗器械消毒剂、与透析机配合使用的专用水处理设备,以及与麻醉机配合使用的气瓶/减压装置。
还需要指出的是,定制设备通常是根据医疗专业人员的书面处方为特定个人专门制造的独特器械。牙科器具、假体和助听器插入件是定制器械的一些示例。牙科合金、牙科陶瓷以及假体用模块化组件则属于特定用途定制设备的一些实例。还应注意,某些产品尽管外观类似医疗器械,但若非用于“医疗用途”,则不被视为医疗器械。示例包括:
- 婴儿尿布、女性卫生用品(卫生巾、卫生棉条)、吸奶器、牙刷、牙签、牙线、牙齿美白/漂白产品、床垫保护套和手部清洁湿巾;
- 用于保护使用者的口罩(例如,防止环境危害)不属于医疗器械。然而,外科口罩(在手术室中使用)属于医疗器械,因为其目的是保护患者而非使用者。护齿器仅在用于特定“医疗”目的时才属于医疗器械,例如正畸治疗后的保持器;
- 乳胶/橡胶手套——检查手套和外科手套属于医疗器械。用于其他目的的手套不属于器械(例如,用于家庭或实验室中)。根据医疗器械法规,以主要用于医疗器械消毒的消毒剂不得附带多用途使用的声明;因此,此类产品只有在预期用于医疗器械时才可加贴 CE标志。
15.3 医疗器械生命周期阶段
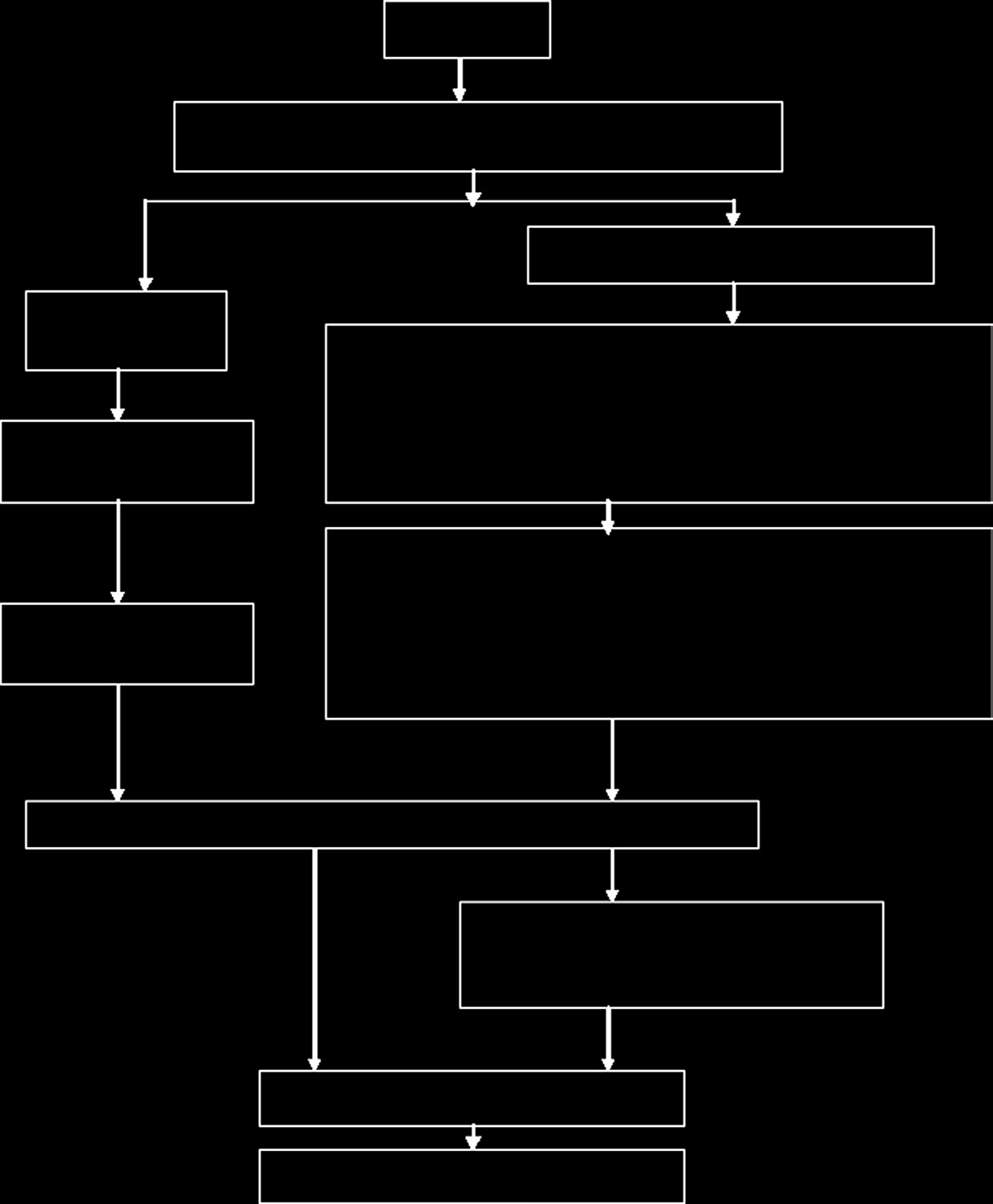
医疗器械的生命周期阶段如图15.2所示,从医疗器械的概念设计到其处置[10]。
这些活动阶段已被简化,以便于理解监管体系。例如,开发阶段包括开发规划、设计验证/确认、原型测试和临床试验。
医疗器械的生命周期各阶段由不同的人员控制。一般情况下,生产商负责并管理医疗器械生命周期的前三个阶段,即从产品概念到设计阶段。医疗器械供应商则负责医疗器械生命周期的第二部分,即从广告到销售阶段。
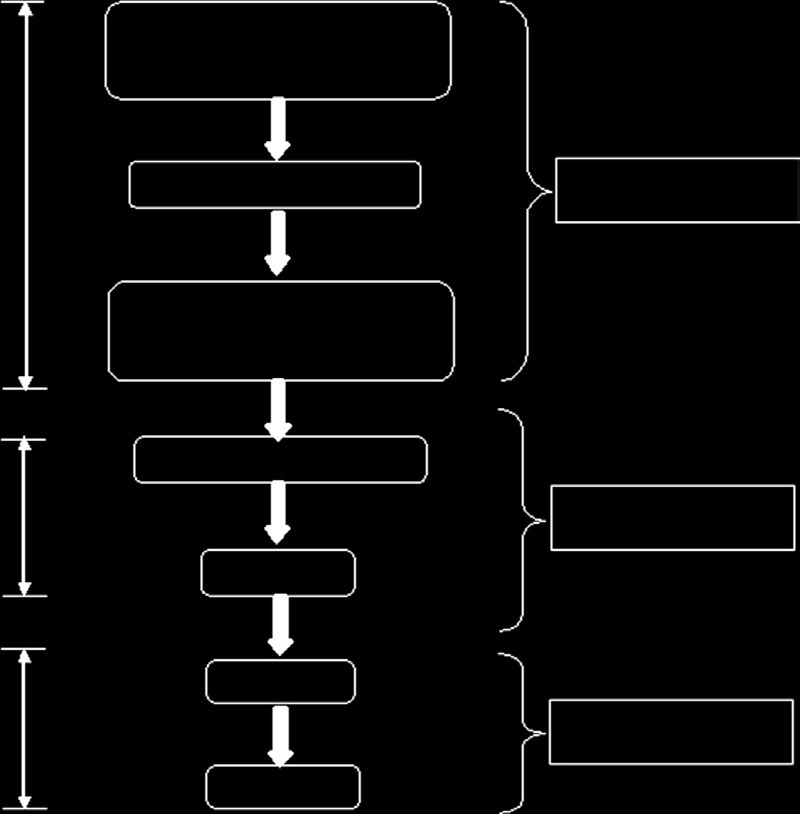
表15.2 医疗器械监管的通用框架 [10]
| 阶段 | 控制/监控 | 人员 | 受监管活动 |
|---|---|---|---|
| 上市前 | 产品 | 制造商 | 器械属性–安全性和制造性能–质量体系 标签–准确的描述‐ 该产品的形成和清晰度 使用说明 |
| 上市 | 市场销售 | 供应商 | 机构注册 – List可用产品的和 法规要求供应商履行售后义务 Advertising–禁止误导‐ 或欺诈行为 |
| 售后/使用 | 售后/使用 | 供应商/用户 | 监控–售后义务,临床性能的器械和问题识别 问题识别 |
除了上述控制因素外,政府实施的法规还对医疗器械的不同阶段进行管控。
根据政府政策,医疗器械的生命周期 broadly 分为三个部分,即上市前控制、上市后监管与控制以及上市后监测。上市前控制阶段通过满足国家的法规要求,确保开发的医疗器械产品正确进入市场。标签和广告控制用于确保产品的正确表述。上市控制确保机构注册、器械列名和售后义务的落实。上市后监测/警戒则确保使用中器械的持续安全性和性能。
15.4 欧洲医疗器械指令法规
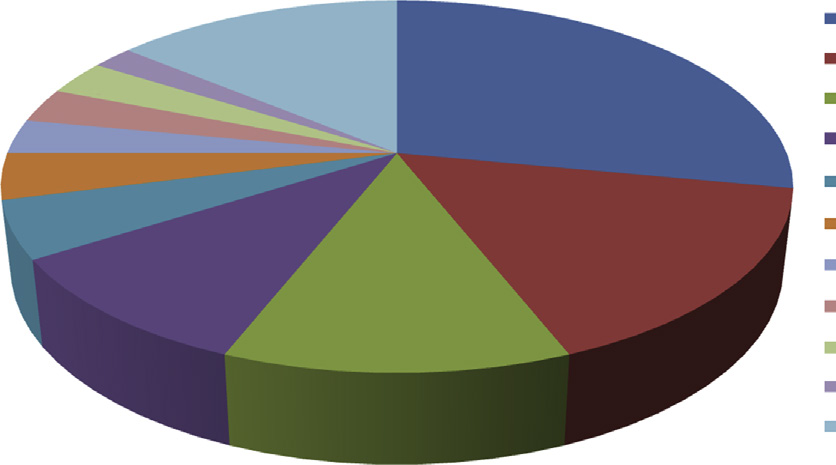
欧洲医疗器械市场是全球第二大医疗器械市场,估值约为1000亿欧元 [11]。仅次于美国市场,欧洲市场约占全球医疗器械市场的31% [12]。2014年,约有1.1万件专利申请提交至欧洲专利局的医疗技术领域,占总申请量的7%,数量巨大。其中,41%来自欧洲国家(28个欧盟成员国、挪威和瑞士),59%来自其他国家 [13]。在欧洲市场中,德国、法国、英国、意大利和西班牙是欧洲最大的医用纺织品市场。这些国家也是欧洲体外诊断设备(IVD)市场的主要(前五位)贡献者 [14]。 图15.3 显示了欧盟的医疗器械市场份额。
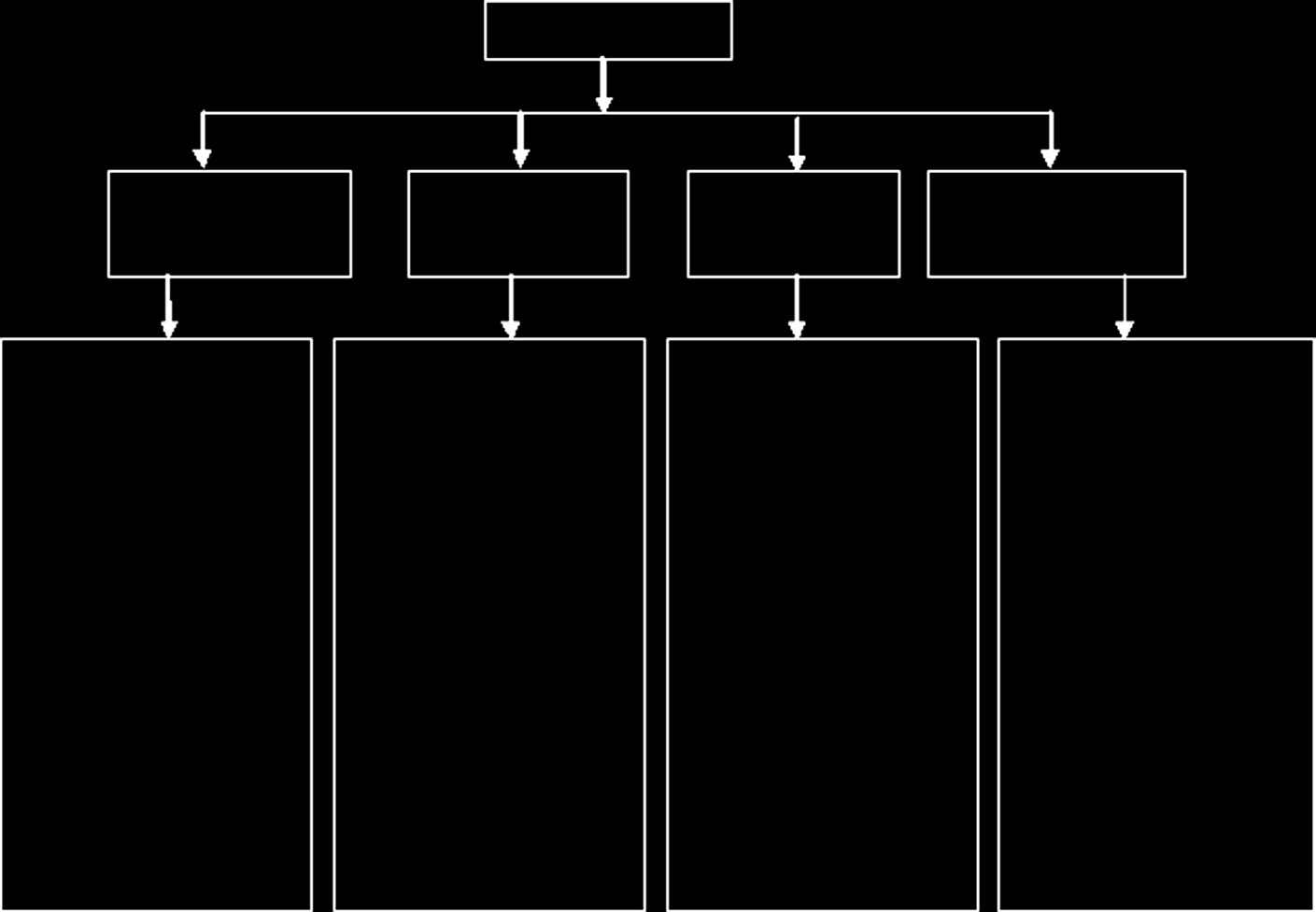
医疗器械指令(MDD)自1990年代起规范了欧洲医疗器械的安全性和上市销售。医疗器械根据器械使用相关的风险程度、器械与人体接触的时间以及器械的侵入程度进行分类。与美国不同,欧盟采用以下分类:
市场份额百分比
14 3 3 3 4 5
10 12
16
28 2
德国 法国 UK 意大利 西班牙 荷兰 Swiz 瑞典 比利时 奥地利 其他
- I类:低风险器械,如手套、绷带、听诊器、病床、轮椅及其他患者支持系统;
- IIa类:中等风险(低至中等)器械,如助听器、心电图机、超声诊断设备及其他电子医疗设备;
- IIb类:较高风险(中至高)器械,如手术激光器、输液泵(非植入式)、呼吸机、重症监护监测设备等;以及
- III类:最高风险器械,如导管、人工心脏瓣膜。
医疗器械在欧盟市场销售必须遵守严格的医疗器械指令(MDD)法规。其中一项规定是加贴CE标志 [15]。自1998年6月14日起,所有受MDD 93/42/EEC覆盖的医疗器械,若无CE标志,则不得投放市场。唯一不需要CE标志的器械是“定制器械”和“用于临床研究的器械”,但制造商必须按照 MDD要求保留相关文件。
15.4.1 医疗器械分类
医疗器械分为I类、II类(IIa和IIb)以及III类器械。II类器械所需的评估级别将高于I类器械。对于III类器械,其评估标准将比II类器械更为严格。分类规则在指令的附件IX中规定。该附件包含了分类规则中所用术语的定义。医疗器械的分类将取决于一系列因素,包括:
- 器械的预期使用时长
- 器械的性质,该器械是否为侵入性/外科侵入性。
- 器械的使用方式,它是可植入的还是有源的?
- 该器械是否含有物质?该物质本身被视为药用物质,并且其作用是作为器械作用的辅助。
通常是“预期用途”决定医疗器械的类别,而不是器械的特定技术特性。是由制造商为器械指定的“预期用途”来确定医疗器械的类别,而不是由同一制造商或其他制造商生产的其他类似产品所分配的类别[16]。
15.4.2 分类规则的使用方式
器械的分类基于随器械提供的信息中的声明来确定。制造商必须充分具体地说明器械。制造商必须提供使用所需的最低要求,或者适当的阳性或阴性指征 [17]。
根据任何分类规则,若某器械被列为“特定用途”,则该产品制造商应明确指出,该产品或医疗器械在与器械相关的信息中拟用于此类特定目的。否则,该器械可能会被视为主要用于一般医疗实践的普通器械,即其主要设计和使用目的。此类器械的示例
是激光打印机和识别摄像头,这些设备可与医疗器械结合使用。只有当制造商以特定预期用途将这些器械投放市场时,它们才会被视为医疗器械。
指令和相关法规的附件IX中列出了18条规则,这些规则规定了分类的基本原则。在MEDDEV 2.4/1 第8版中,这些规则得到了进一步解释,并提供了描述性示例。这18条规则如[17]表15.3所示,分为4组: 需要强调的是,在对某些器械进行分类时存在实际问题,例如,伤口敷料根据实际使用方式被划分为I类、IIa类或IIb类。
- 预期用作机械屏障——I类(吸收垫、创可贴和邦迪)。这些敷料起到屏障作用,保持伤口位置或吸收伤口渗出物。
- 用于伤口微环境——IIa类:具有特定附加愈合性能的器械。例如,旨在管理伤口床湿度但不用于需通过二期愈合的大面积伤口的敷料。
- 用于暴露伤口——IIb类:通过二期愈合方式愈合的伤口,如肉芽形成。
- 用于腔隙性伤口、烧伤伤口和慢性溃疡伤口的敷料为IIb类。
15.4.3 合规要求
有三项与医疗器械相关的指令 [18,19]:
- 医疗器械指令(MDD)– 93/42/EEC
- 有源植入式医疗器械指令(AIMD指令)– 90/385/EEC
- 体外诊断医疗器械指令(IVDD指令)– 98/79/EC。
指令90/385/EWG(AIMD)和93/42/EC(MDD)已由欧洲议会和2007年9月5日理事会通过的指令2007/47/EC进行了变更。这些变更自2010年3月21日起实施。以下阐述主要针对MDD,尽管其他两项指令中的大部分规定基本相同。
- 医疗器械指令(MDD)(93/42/EEC 和 2007/47/EEC):适用于所有未被有源植入式医疗器械指令或体外诊断医疗器械以及人源和动物源产品所涵盖的一般医疗器械。一般医疗器械包括从简单的绷带到高科技放射设备。
- 有源植入式医疗器械指令(AIMDD)(90/385/EEC 和 2007/47/EEC):适用于所有预期永久植入人体的有源设备及相关附件。这包括所有植入并留置于人体内的有源医疗器械,例如起搏器、植入式除颤器、植入式输液泵、植入式神经肌肉刺激器和膀胱刺激器
- 体外诊断医疗器械指令(IVDD)(98/79/EEC):适用于所有在远离患者的地方用于诊断患者医疗状况的设备和套件。这包括任何用于体外检查源自人体的物质的医疗器械、试剂、试剂产品套件、仪器、装置[18]。
这些指令的主要目的是确保器械的安全性和性能,并允许医疗器械在欧盟内的自由流通。医疗器械指令(MDD)的十二个附录定义了医疗器械的合规要求[20]:
- 附录I – 基本要求,包括通用要求和设计和构造要求。通用规定指出,医疗器械的设计和开发应确保在预期条件和用途下使用时,不会损害患者、用户或其他人员的健康或安全。安全、运输和储存条件也包含在本附录中。设计和构造要求附录涵盖的要求包括化学、物理和生物学特性(如感染和微生物污染)、辐射防护、电、机械、热风险、能源供应或能量物质以及标签要求和使用说明。
- 以下列出了 附录II:欧共体符合性声明(全面质量保证体系)中的一些关键要求: 制造商质量体系必须注册符合适用的 ISO 9000 和 EN46000 标准,并接受常规监督审核。制造商必须声明其符合医疗器械指令(MDD)的要求,并按照第17条和附录XII的规定加贴 CE 标志。制造商需要在最后一批产品制造完成后至少保留其符合性声明5年。
- 附录III: 欧洲类型检验证书:要求公告机构对器械的代表性样品进行测试和评估,以确保该器械完全符合医疗器械指令(MDD)的适用要求以及适当的技术标准。
- 附录IV: 欧洲共同体验证:包括欧洲共同体验证符合性,要求医疗器械制造商(或其授权代表)声明该产品符合所有适用的 MDD 要求和相应的技术规范。
- 符合 附录V 的要求必须结合附录III 或 附录VII 中规定的程序(根据产品分类而定)。
- 附录VI: 欧共体符合性声明(产品质量保证):制造商必须依据适当的 ISO 9000 或 EN46000 获得产品质量保证注册(仅限最终检验)。
- 附录VII: 欧共体符合性声明:制造商需要准备适当的技术文档,以证明其完全符合该指令及相关技术标准的要求。
- 附录VIII:关于特殊用途器械的声明:制造商必须提供与定制型器械或用于临床研究的器械的预期用途相关的特定文件。
- 附件IX:分类标准:包括定义、实施规则的程序以及分类规则本身。
- 附录X: 临床评价:适用于拟用于临床研究的器械的相关要求。这些要求包括数据的汇编、保密性声明和文件
- 附录XI: 指定公告机构需满足的标准:定义了被指定为公告机构的组织的选择标准、行为和责任。
- 附录XII: 符合性CE标志:定义了CE标志的物理尺寸和外观。
15.4.4 CE标志
根据欧盟MDD的三种分类之一涵盖的医疗器械,在投放市场前必须加贴CE标志。CE标志是制造商发布的声明,表明该产品符合相关欧洲指令的要求。CE标志使企业能够更便捷地进入欧盟和欧洲自由贸易区市场销售其产品,而无需进行适应性修改或重新检查。与CE标志相关的一些显著特征如下:
- 器械制造商/代理人应贴附CE标志。
- 所贴附的CE标志必须清晰可见、易读且持久。
- 必须随CE符合性标志一并提供通知机构的识别编号。
- 如果缩小或放大CE标志,必须遵守上述比例图示中的比例。当相关指令未规定具体尺寸时,CE标志的高度不得小于5 mm。
- 欧洲31国(28个欧盟成员国加上欧洲经济区条约的3个签署国——冰岛、列支敦士登和挪威)需要CE标志。瑞士不属于欧盟/欧洲经济区,但已将欧盟MDD纳入本国法律。
- CE标志并不保证产品的质量,但它认证该器械符合基本要求,并向用户保证产品能够按照制造商的预期功能正常运行,且在按预期使用时是安全的。
医疗器械上市销售流程图如图15.4所示。
15.4.5 公告机构
公告机构(NB)是一个独立的审查机构,由各国相应的监管机构(卫生部)授权,在指令范围内执行符合性评估活动。英国的NB示例包括英国标准协会、Amtac认证服务有限公司、天祥检测与认证有限公司、劳氏质量保证有限公司、SGS英国有限公司、UL国际(英国)有限公司。欧盟NB名单会定期在《欧洲共同体官方期刊》上发布,列于93/42/EEC指令下通报机构名单中。
公告机构将对中等和高风险器械(IIa类、IIb类和III类)进行评估,而不评估低风险器械。高风险IIb类和III类器械需在实验测试环境中由公告机构进行评估。公告机构的部分服务如下:
- 产品测试
- 检验证书颁发
必须注意,制造商自身需对I类低风险器械的生产过程和产品本身评估承担全面责任。公告机构在对产品进行评估时,必须保持判断的公正性,不得参与所评估产品的设计、制造、维护或分销。
15.4.6 技术文件
制造商必须准备一份技术文件,以证明该产品符合医疗器械指令并满足基本要求。所有三类器械都需要技术文件。术语“技术文件”适用于I类、IIa类和 IIb类器械,而“设计档案”适用于第三类医疗器械。通常,技术文件或设计档案包含12个部分,如表15.4所示。
技术文件由公告机构进行审核。在产品加贴CE标志之前,必须将设计档案提交公告机构进行审查。成功通过审查后,公告机构将根据理事会指令附录 II.4颁发设计审查证书,证明该产品符合医疗器械指令附录I的相关规定。
15.4.7 符合性评估程序
符合性评估程序用于证明器械符合93/42/EEC指令的要求。合规性是通过建立符合性声明而形成的一项法律约束文件。合规文件
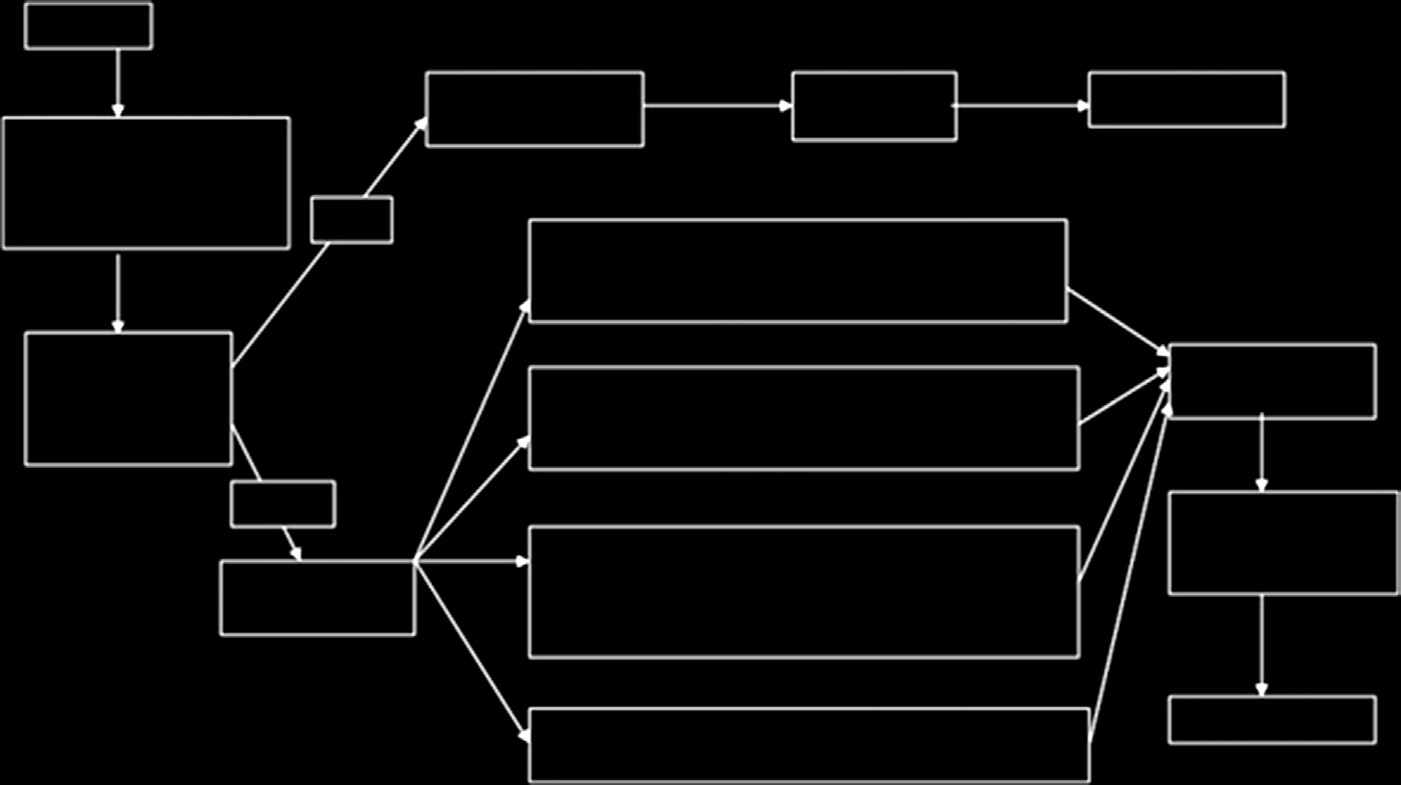
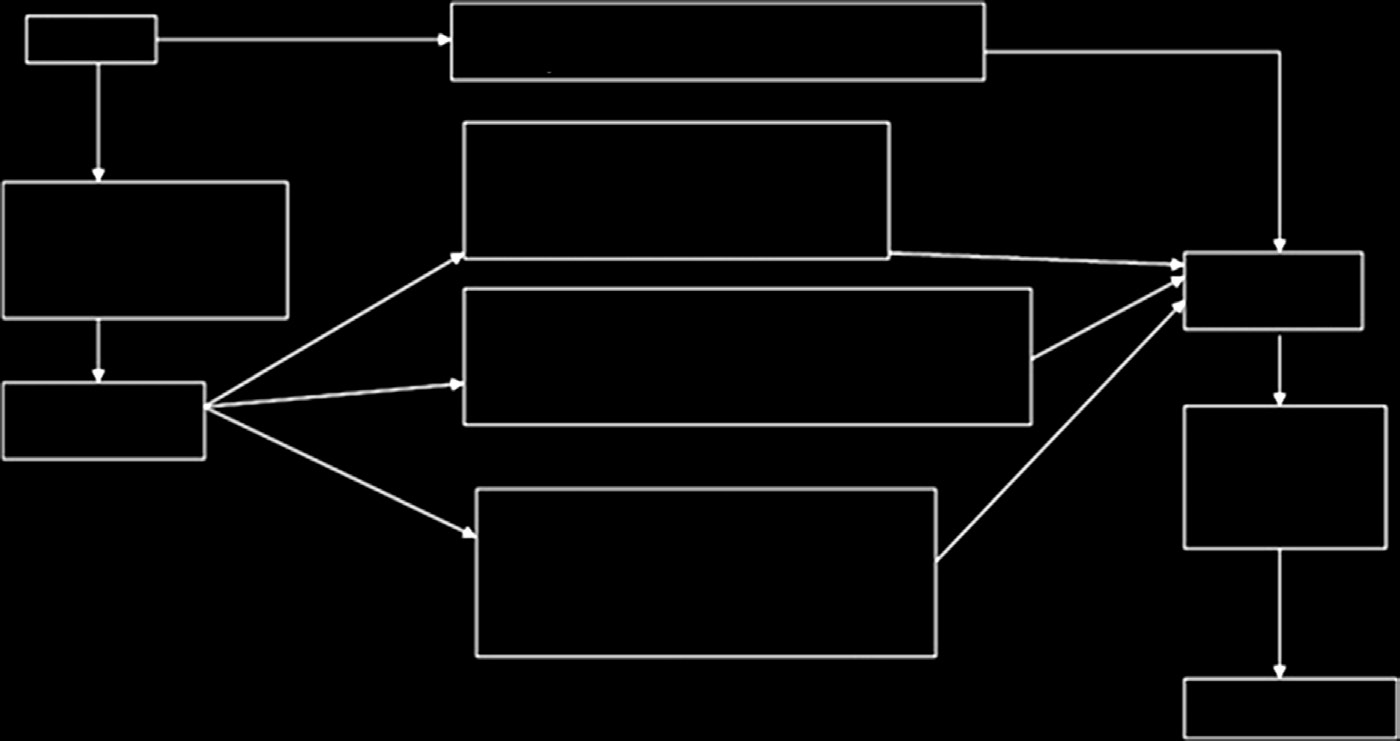
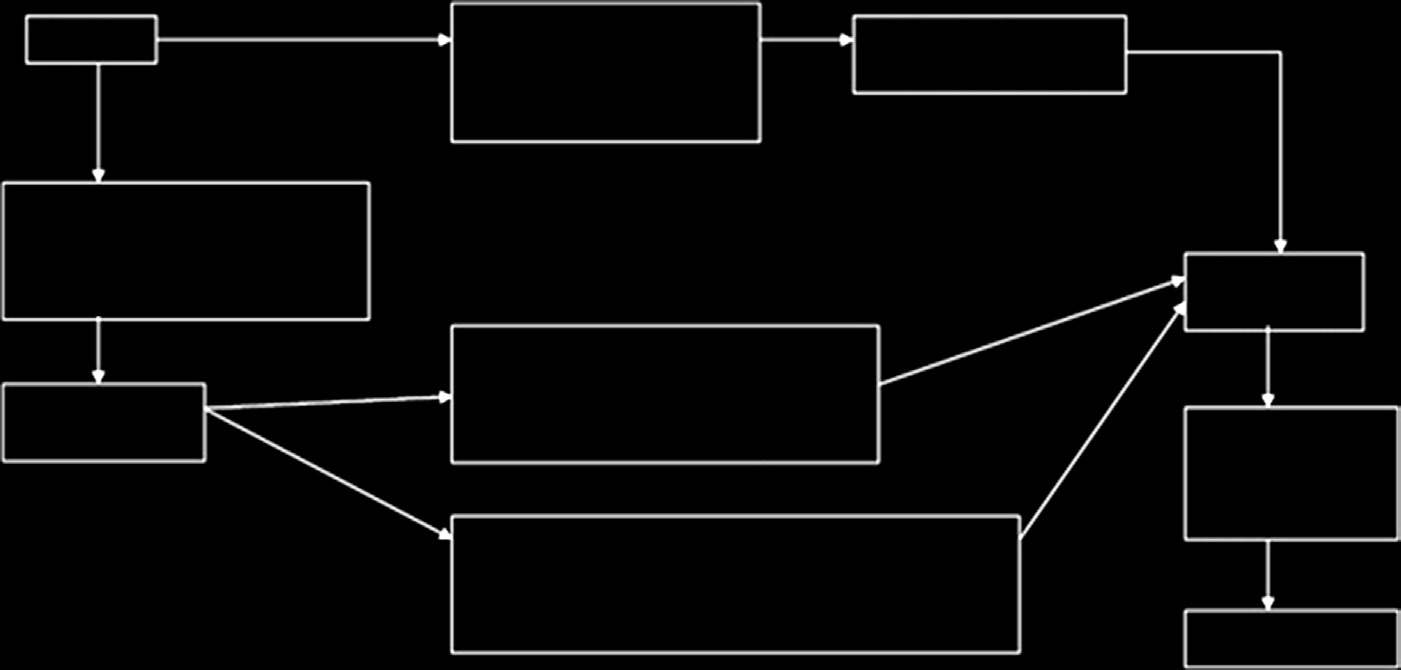
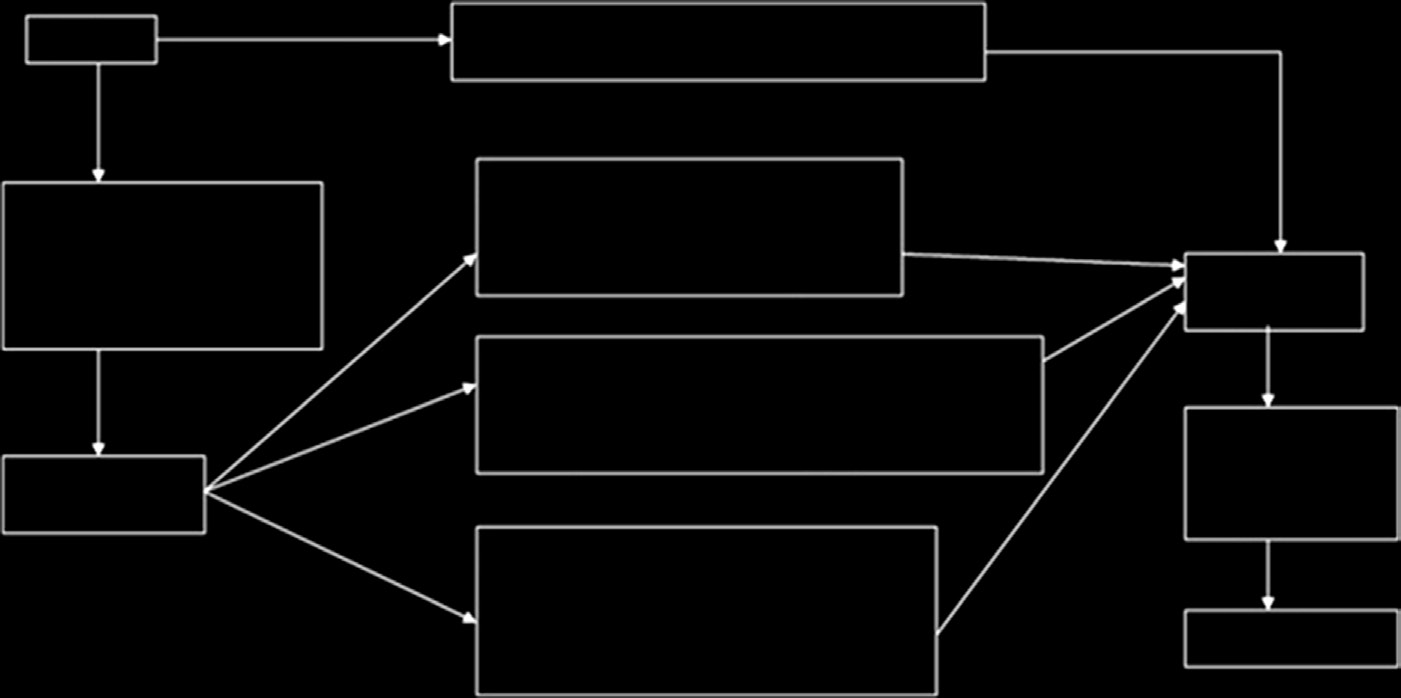
必须包含必要且充分的信息,以便从用户追溯到产品的制造商或欧盟代表。其中可包括该产品符合的指令和标准清单、产品识别信息以及制造商的名称、地址和签名。必须由公告机构颁发的合格证书进行验证。I类、IIa类、IIb类和 III类医疗器械的符合性评估程序分别如图15.5–15.8所示[15]。
I类器械不需要接受质量体系的审核,技术文件也无需由公告机构审核。相反,必须在符合性声明中进行声明。全面的质量体系适用于IIa类、IIb类和III类医疗器械。
15.4.8 伤口敷料和法规规范
如前几节所述,伤口敷料也被归类为医疗器械,并受欧洲MDD的监管。欧洲标准EN‐13795涵盖了MDD中所有缺失的技术部分,包括产品从设计、开发和生产到其安全使用条件、储存和包装(包括运输、标签)以及其他医疗器械的理化和微生物特性等方面。
表15.4 设计档案或技术文件的不同部分
| 内容编号 | 内容详情 |
|---|---|
| 1. | 引言 |
| 2. | 基本要求检查清单 |
| 3. | 风险分析 |
| 4. | 图纸、设计、产品规格 |
| 5. | 化学、物理和生物测试 |
| 5.1 | 体外测试——临床前研究 |
| 5.2 | 生物相容性测试 |
| 5.3 | 生物稳定性测试 |
| 5.4 | 微生物安全性、动物源组织 |
| 5.5 | 涂层医疗器械 |
| 6. | 临床数据 |
| 7. | 包装验证和保质期 |
| 8. | 标签 |
| • | 使用说明 |
| • | 患者信息 |
| • | 宣传材料 |
| 9. | 制造 |
| 10. | 灭菌 |
| 11. | 结论 |
| 12. | 符合性声明(草案) |
| ## 15.4.8.1 I类伤口敷料 |
根据分类规则1,所有非侵入性设备均属于I类。
例如:用于辅助扭伤愈合的非无菌敷料、石膏、颈托、重力牵引装置、压力袜。
所有与受损皮肤接触的非侵入性器械均属于I类。但是,如果其预期用途为机械屏障、压迫或渗出液吸收,则适用规则4。
例如:吸收垫、岛状敷料、药棉、伤口条带、粘性绷带(创可贴、邦迪)以及作为屏障、保持伤口位置或吸收伤口渗出物的纱布敷料。
15.4.8.2 IIa类伤口敷料
所有与受损皮肤接触的非侵入性器械均属于IIa类,但包括主要用于管理伤口微环境的器械在内的敷料,应归入分类规则4。
旨在通过在愈合过程中控制伤口处的水分水平(湿性愈合理论)以及通常调节湿度和温度环境来辅助愈合过程的敷料,
并通过其他物理方式影响氧气和其他气体以及pH值水平或影响该过程的敷料,也被归类为IIa类外器械用。粘合剂、聚合物薄膜敷料–水凝胶敷料–非药物浸渍纱布敷料。
15.4.8.3 IIb类伤口敷料
根据分类规则4,所有与受损皮肤接触的非侵入性器械,如果主要用于真皮层已破损且只能通过二期愈合方式愈合的伤口,则属于IIb类。
用于慢性广泛溃疡性伤口的敷料–用于严重烧伤且已穿透真皮并覆盖大面积区域的敷料–用于严重压疮伤口的敷料–含有促进组织生长功能并提供临时性皮肤替代物的敷料
15.4.8.4 III类伤口敷料
- 所有包含一种物质作为其组成部分的器械,如果该物质单独使用时可被视为符合 2001/83/EC指令第1条定义的药品,并且该物质在人体上的作用对器械的主要作用起辅助作用,则根据分类规则13归为III类。例如:含有抗菌剂的敷料,其中该抗菌剂的作用是对伤口提供辅助作用
- 所有利用动物组织或无生命活性的衍生物制造的器械,若预期与完整皮肤接触除外,均属于III类,依据规则17。例如:由胶原蛋白制成的植入物和敷料。
15.5 美国医疗器械法规
在美国,大多数医疗器械产业集群位于加利福尼亚州、马萨诸塞州、纽约州和明尼苏达州地区。仅在美国就有超过7000家医疗器械公司,年销售额约为每年 1100亿至1160亿美元[21]。美国医疗器械市场是全球最大的市场,2016年估计达到1477亿美元。美国医疗器械市场预计在未来3年的复合年均增长率为6.1% [22]。美国医疗器械行业直接雇佣40万名美国人,间接雇佣200万外国人[23]。
15.5.1 上市前审查程序
负责监管美国医疗器械的联邦机构是食品药品管理局(FDA)——隶属于卫生与公共服务部的一个机构。制造商必须获得美国食品药品监督管理局的事先批准或许可
在美国上市销售许多医疗器械之前。美国食品药品监督管理局(FDA)的器械与放射健康中心(CDRH)主要负责医疗器械上市前审查。然而,与血液采集和处理程序、细胞产品和组织相关的器械则由生物制品评价与研究中心监管[24]。
医疗器械制造商必须向美国食品药品监督管理局注册其设施并列明其设备,同时遵守一般监管要求。美国食品药品监督管理局根据医疗器械对消费者带来的风险对其进行分类。低风险医疗器械如塑料绷带和冰袋,对用户不构成风险。
此类器械在完成法定注册后即可直接上市销售,无需经过美国食品药品监督管理局的任何监管程序。但是,中高风险的器械在上市前必须经过美国食品药品监督管理局的全部监管程序。只有当产品符合所有法规要求(包括上市前要求和必要的上市后要求)时,美国食品药品监督管理局才会向制造商授予批准。
15.5.2 联邦法规法典
联邦法规法典(CFR)是联邦政府行政部门和机构在联邦公报中发布的通用和永久性规则的汇编[25]。美国食品药品监督管理局(FDA)的法规列在联邦法规法典第21篇中。联邦法规法典的每一标题(或卷)每年修订一次。修订后的第21篇大约于每年4月1日发布。医疗器械准入所使用的主要部分如下所示[26],
- 21联邦法规法典 第800部分 – 一般规定
- 21联邦法规法典 第801部分 – 标签
- 21联邦法规法典 第803部分 – 医疗器械报告
- 21联邦法规法典 第807部分 – 机构注册和器械列名
- 21联邦法规法典 第808部分 – 州和地方医疗器械要求的联邦优先权豁免
- 21联邦法规法典 第809部分 – IN VITRO 人类使用体外诊断产品
- 21联邦法规法典 第810部分 – 医疗器械召回权限
- 21联邦法规法典 第812部分 – 研究性器械豁免
- 21联邦法规法典 第814部分 – 医疗器械上市前批准
- 21联邦法规法典 第820部分 – 质量体系法规
- 21联邦法规法典 第821部分 – 医疗器械追踪要求
- 21联邦法规法典 第822部分 – 上市后监督
- 21联邦法规法典 第830部分 – 唯一器械标识
美国食品药品监督管理局将1700多种不同的医疗器械分类并归入医学专业 ‘小组’。这些小组记录在《联邦法规法典》第21篇(21 CFR)第862–892 部分中。一般来说,如果医疗器械制造商的产品属于中高风险器械类别,则有两种不同的途径可在获得美国食品药品监督管理局许可的情况下将其器械引入市场[27路]。径1:开展临床研究,提交上市前批准(PMA)申请,并提供合理保证器械安全有效的证据。PMA流程通常用于新型高风险器械,并获得美国食品药品监督管理局称为批准的一种许可类型。
路径2:在此流程中,需要提交510(k)通知,通过提及并声明市场上已存在的一种不需要PMA的实质等效器械(同品种器械)。这是一种相对便宜且较短的途径。该路径更短且成本更低。510(k)流程是医疗器械特有的,结果为 FDA clearance(许可)。实质性等效是通过比较新器械与同品种器械的性能特性来确定的。
15.5.3 器械分类
根据1976年医疗器械修正案(MDA,公法94‐295)的规定,美国食品药品监督管理局将当时在市场上销售的所有医疗器械——即修正案前的器械,分为三类之一。国会根据风险(低、中等
和器械对患者构成的高风险)的分类。每类的示例列于表15.5中。
I类 – 根据现行法规,I类器械是指能够充分提供器械安全性和有效性的合理保证的器械。许多I类器械免于上市前通知和/或质量体系法规要求 [27]。
II类 – II类包括对患者具有中等风险的器械,可能包含有可用信息或特殊控制以降低或缓解风险的新器械。特殊控制通常针对特定器械,可能包括特殊标签要求、强制性性能标准和上市后监测。
III类 –这些器械“被宣称用于支持或维持人类生命,或用于在预防人类健康损害方面具有重大重要性的用途”,或“存在不合理疾病或损伤风险,需接受上市前批准以提供安全性和有效性的合理保证”。[27].
表15.5 美国食品药品监督管理局对医疗器械的分类
| 器械分类 | 示例 | 安全控制 | 提交要求 |
|---|---|---|---|
| I类 | 弹性绷带,examination手套,手持手术器械 | 一般控制 | 仅需注册,510(k)豁免 |
| II类 | 电动轮椅,输液泵,手术窗帘 | 一般控制和特殊控制 | 510(k)通知,除非豁免 |
| III类 | 心脏瓣膜、硅胶乳房植入物,植入式小脑stimulator | 一般控制和上市前批准 | PMA申请,IDE可能 |
| 金属对金属髋关节, 某些牙科植入物 | 一般控制 | 510(k)通知 |
15.5.4 上市前批准
上市前批准是用于全新或高风险器械的审批方法之一,也是美国食品药品监督管理局对器械审批流程中最严格的手段之一。通过PMA系统对该产品的批准主要基于有效科学证据,以合理保证该器械在其申请中所述预期用途下的安全有效。与510(k)相比,PMA通常要求在获得美国食品药品监督管理局批准前提供一些临床数据[28]。
医疗器械在启动临床研究之前必须获得研究性器械豁免(IDE)。IDE流程允许将未经批准的器械(最常见的是侵入性或维持生命的器械)用于临床研究,以收集支持上市前批准(PMA)申请所需的数据[29]。PMA必须包含(除其他内容外)以下信息:
- 支持该使用方式的非临床和临床数据摘要以及从研究中得出的结论;
- 包含重要物理和性能特性的器械描述;
- 预期用途,患者人群的描述以及该器械将用于诊断、治疗、预防、治愈或缓解的疾病或状况;
- 国内外销售历史的描述,包括该器械是否因与器械安全性和有效性相关的任何原因而被撤市;
- 拟定的标签;以及生产过程的描述
15.5.5 上市前批准补充申请
如果制造商希望对已批准的PMA器械进行修改,必须向美国食品药品监督管理局提交几种不同类型的上市前批准补充申请之一。各种类型的上市前批准补充申请在表156[27,30]中简要描述。制造商还需支付用户费用,但特殊上市前批准补充申请除外。美国食品药品监督管理局在其网站上提供有关已批准的上市前批准补充申请的信息。
表15.6 美国不同的PMA补充程序
| 类型 | 补充 | 变更类型 | 到器械 | 需要的数据 | 评审员 | Year | 引入 |
|---|---|---|---|---|---|---|---|
| 面板跟踪 | 重大设计变更;新适应症 | 临床,有限临床前数据 | 在某些情况下 | 专题领域专家组和/或 FDA工作人员 | 1990 | 180天 | |
| 重大 | 重大设计变更;标签变更 | 临床前;临床确认性数据 | 在某些情况下 | FDA工作人员 | 1986 | 180天 | |
| 实时 | 轻微设计变更至器械,软件,灭菌或标签 | 仅临床前 | FDA工作人员 | 1997 | 30天通知 | ||
| 特殊 | 制造变更 | 无特定数据需要 | FDA工作人员 | 1997 | |||
| 标签变更增强设备安全 | 无特定数据需要 | FDA工作人员 | 1986 |
15.5.6 510(k)通知
对于中等风险医疗器械,若其不豁免上市前审查程序,则通常需要进行 510(k)提交。该510(k)也可用于当前已上市的器械,当制造商寻求新的适应症(例如新人群,如儿科使用,或新疾病或状况),或对器械的设计或技术特性进行了可能影响其性能特性的变更时。PMA和510(k)审批途径在审查流程上存在根本性差异。在PMA审查中,美国食品药品监督管理局会确定器械的安全性和有效性;而在510(k)审查中,美国食品药品监督管理局则判断该器械是否与已在市场上销售的器械实质等同。但此前可能并未对现有器械的安全性和有效性的评估[27,31]:
- 510(k) 应当提交
- 将新器械引入市场
- 更改先前获批器械的使用适应症
- 以及对先前获批器械进行重大调节。
- 510(k)是必须的
- 制造商
- 规范制定者
- 对器械或其标签进行变更的重新包装者
- 以及同时制造和分销医疗器械的任何人。
- 510(k) 不需要
- 私人标签分销商
- 未对器械或其标签进行变更的重新包装者
- 分销商或进口商,进一步推广合法上市的器械且未更改标签或器械。
510(k)提交和分类流程分别如图15.11和图15.12所示。510(k)与上市前批准程序(PMA)之间的区别见表15.7。
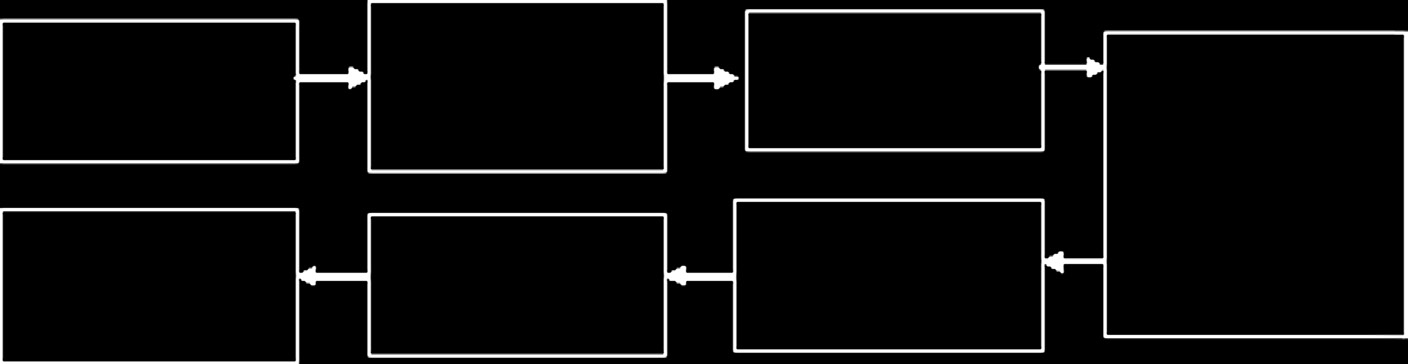
15.5.7 医疗器械审评流程:上市后要求
一旦获得批准或准予上市销售,医疗器械制造商必须遵守有关标签和广告、制造以及上市后监控的各种法规。根据CDRH报告,“当前美国医疗器械上市后监管体系主要依靠”以下来源来发现医疗器械的潜在问题[27]:
- 医疗器械报告(MDR)
- 药品安全网络
- 批准后研究
- 上市后监测研究
- FDA自由裁量研究
- 以及其他工具。
上述有关医疗器械上市后问题的信息来源存在一定的局限性。
15.5.8 美国伤口敷料相关法规
根据美国联邦法规第21章800条,以下是I类、II类和III类伤口敷料器械的一些示例。
表15.7 上市前批准与510(k)流程的区别
| PMA流程 | 510(k)流程 |
|---|---|
| 安全性和有效性 | 实质等同 |
| III类医疗器械 | I类医疗器械 , II类和部分III类 |
| 科学证据 | 与已上市器械的对比 |
| 始终需要临床数据 | 10%–15%的使用方式要求临床数据 |
| 详细且冗长的申请 | 简短的申请流程 |
| 产品将被批准 | 将获得许可 |
| 时间线:180天 | 时间线:传统和简略的: 90天特殊510(k):30天 |
| 批准前检查 | 无需批准前检查 |
| 上市后活动(补充)要求 | 无上市后活动 |
| 不易复制 | 易于复制 |
| 需要咨询小组审查,但不适用于所有PMA | 罕见的咨询小组审查 |
15.5.8.1 I类伤口敷料
简单且侵入性最小的织物敷料被指定为I类器械。根据联邦法规法典,伤口敷料归类于第21篇联邦法规法典第878部分,一般规定和整形手术器械,E小节——手术器械[32]。
- 根据878.4018——亲水性伤口敷料被归类为I类(一般控制)。
- 根据878.4020——封闭性伤口敷料被归类为I类(一般控制),这些器械免于第807部分的上市前通知程序。
- 根据21联邦法规法典第880部分——普通医院和个人使用器械,绷带被列为:
- 880.5090 液体绷带 – I类(一般控制)
- 880.5075 弹性绷带 – I类(一般控制)
- 880.5240 医用胶带和粘性绷带 – I类(一般控制)
- 878.4014 用于体外的不可吸收纱布/海绵。
- 878.4450 用于体内的不可吸收纱布。
15.5.8.2 II类伤口敷料
医用纺织品/伤口敷料,若用作非植入式器械(例如由纤维或织物制成的敷料或贴片形式的抗菌剂)或用作可植入器械(例如心血管移植物),则被归类为II类。原因是当抗菌剂应用于纤维或织物(纺织品)时,被视为药物,而包含该抗菌药物的器械将被视为组合产品,如EN 13795[33]中21 CFR 3.29(e)所定义。
- 含聚(二烯丙基二甲基氯化铵)添加剂的伤口敷料在878.4015中被归类为II类(特殊控制)。
- 878.4011 用于辅助伤口闭合的组织粘合剂——一种用于皮肤表面接近的带有辅助伤口闭合器械的组织粘合剂。
15.5.8.3 III类伤口敷料
含有人体或动物源性生物材料的产品属于III类。III类敷料的示例如下
- 878.4010 用于非体表用途的组织粘合剂。该敷料用于内部组织和血管的粘合。
- 878.4490 可吸收止血剂和敷料——该敷料的预期用途是通过加速血液凝固过程以实现止血。
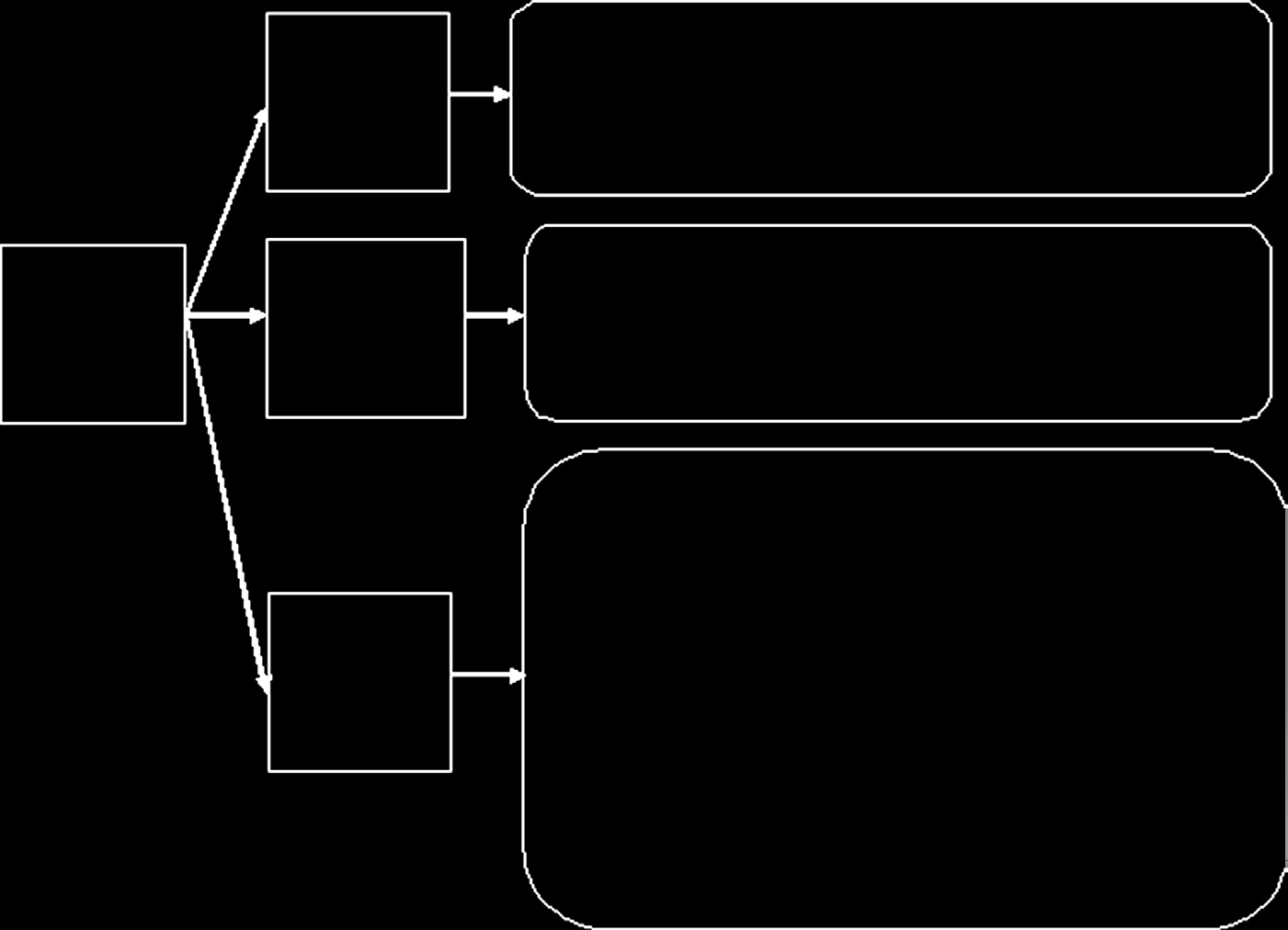
表15.8展示了与不同伤口敷料相关的监管路径。美国医疗器械审批流程的流程图如图15.13所示。
表15.8 伤口护理产品类别 [34]
| 产品类别 | 示例 | 监管路径 |
|---|---|---|
| 传统伤口产品 | 粘性绷带,外用软膏,纱布 | 510k 或 专论 |
| 伤口闭合产品 | 内部缝合器,皮肤缝合器 手术/结扎夹,缝线 | 510k |
| 高级保湿伤口敷料产品 | 藻酸盐敷料,薄膜敷料 泡沫敷料,水胶体敷料,水凝胶敷料 | 510k 或 上市前批准 (PMA)申请 |
| 主动伤口愈合产品 | 基于细胞的皮肤替代物替代,胶原蛋白敷料‐剂,生长因子 | PMA、新药申请或生物制品许可证申请 |
| 清创产品 | 清洁剂和密封剂 | 510k |
| 抗菌敷料 | 含银抗菌敷料,非含银抗菌剂敷料 | 510k 或 上市前批准(PMA) |
15.6 加拿大医疗器械法规
加拿大拥有世界最大的经济体之一,其医疗器械市场排名第八。2015年该市场价值为62亿美元,预计将持续稳定增长,到2020年将达到约86亿美元。医疗器械进口占医疗器械市场的80%。加拿大的医疗总支出占其GDP的10.4%,人均医疗支出为5292美元。[35]。
加拿大的医疗器械由治疗产品局(加拿大卫生部的一个分支)监管,并受《加拿大医疗器械法规》[CMDR(SOR/98‐282)]和《食品药品法》(F& D Act)(R.S., 1985, c. F‐27)的约束。加拿大医疗器械监管在很大程度上结合了欧盟和美国法规的最佳特点。加拿大医疗器械法规根据类似于欧洲法规的风险因素对器械进行分类,有助于确定器械类别并制定针对新兴技术的要求。
制造商的质量体系由第三方进行审查。医疗器械数据库向公众开放,使公众能够识别经监管机构正确许可的器械[36]。
医疗器械(包括体外诊断器械)的制造、销售、广告销售和进口由加拿大卫生部监管,加拿大卫生部是通过《医疗器械法规》对医疗器械进行监管的加拿大机构。制造商必须确保其医疗器械符合《医疗器械法规》(SOR/98‐282,第9条)中规定的安全性和有效性要求。
15.6.1 医疗器械分类
加拿大卫生部医疗器械局还负责医疗器械项目的上市前部分,通过产品评估、器械分类、质量体系和医疗器械许可来监管医疗器械。医疗器械分类根据《加拿大医疗器械法规》附表1中规定的规则分为I类、II类、III类或IV类,其中I类代表最低风险,IV类代表最高风险。针对体外诊断器械有单独的分类规则。表 15.9显示了《加拿大医疗器械法规》。
15.6.2 质量体系
制造商必须向加拿大卫生部提交II类、III类和IV类医疗器械的医疗器械许可证申请。必须提交ISO 13485:2003
表15.9 加拿大医疗器械分类及监管要求 [38]
| 器械类别 | Risk | 示例–非体外诊断医疗器械 | 示例–体外诊断医疗器械 | 要求 | 许可证 |
|---|---|---|---|---|---|
| I类 | 最低 | 手动可重复使用外科手术器械、医院 beds,担架 ,轮‐椅、粘性绷‐带、手术单,细胞培养基 | 微生和物细学胞培养培养基,一般规定诊断试剂 | 医疗器械establish‐ment 许可证 – for man‐ufacturer,进口商 和 分销商 | |
| II类 | Low | 隐形眼镜。endoscopes,水凝胶敷料,血袋,手术钻 | 家用孕检‐南希试剂盒,实验室用阵列 用于葡萄糖,全血计数和治疗药物监控 | 医疗器械许可证 制造商 医疗器械许建可立证– –用于进口商和 分销商 | |
| III类 | 中等 | 骨科、皮肤用和牙科植入物,血液透析机,手术激光器,骨骨水泥 | 床旁血液葡萄糖监测试纸、药物滥用筛查滥用筛查ing,诊断癌症标志物。 | ||
| IV类 | High | 心脏起搏器,血管造影导管,颅内分流管,替换‐置置换心脏瓣膜,闭环治疗系统,心脏缝线 | 传染性疾病制造者用于危及生命的疾病和供体筛查。例如:人类免疫缺陷病毒 E.g.: HIV |
质量体系证书以及许可证申请。医疗器械的许可证由加拿大标准委员会认可的注册官根据加拿大医疗器械符合性评估系统(CMDCAS)[39]颁发。许可证在完成第三方审核,确认符合相关标准、《加拿大医疗器械法规》(CMDR)以及CMDCAS项目的具体要求后签发。此次注册和质量审核的费用须由制造商承担。I类、定制型和研究用器械的制造商免除此项要求。在加拿大销售的所有医疗器械必须满足CMDR的安全性和有效性要求,并且标签标注必须符合法规规定[40]。
15.6.3 标签
根据《医疗器械法规》(SOR/98‐282,第21‐23条),所有进口到加拿大的医疗器械的标签必须“清晰、持久且显著”,并须包含以下最低限度的数据:
- 器械名称
- 制造商名称和地址
- III类和IV类器械的控制编号
- 如果包装不透明,则包装上应包含内容信息
- 对于需以无菌状态出售的器械,应标注“无菌”字样
- 器械的有效期
- 必须提供器械的用途和目的,除非对预期使用者和医疗状况而言是显而易见的
- 储存和使用说明
对于面向公众销售的器械,标签信息应清晰可读,并印制在包装外部,在正常条件下应可见。标签必须使用英语和法语,且在购买时必须提供这两种官方语言的使用说明。
15.6.4 上市后监管
所有类别的医疗器械的制造商和分销商均须遵守加拿大的上市后监管法规。食品部门和健康产品的检查员通过处理强制性问题报告和召回来进行上市后监督活动。制造商和进口商也有责任进行强制性问题报告。强制性问题报告要求 [41]适用于涉及正式销售的器械的任何事件,包括正常或异常情况,例如
- 与器械的故障或停用、其功效下降,或其标签或使用方式的不足有关。
- 该器械已导致或已引起患者、用户或其他人员的健康状况损失或严重恶化,或若再次发生可能造成此类后果。
如果出现死亡等严重不良后果,制造商和进口商必须在10天内向加拿大卫生部提交此报告。对于非严重问题,制造商和进口商应在事发之日起30天内提交强制性问题报告。
15.6.5 加拿大关于伤口敷料的法规
对于常用作机械屏障或用于压迫或渗出液吸收的器械,其法规详见子规则(1),并归类为I类。此外,I类还包括与受损皮肤接触的非侵入性设备[42]。根据医疗器械分类规则4:(i) 在符合子规则(ii)的前提下,所有预期与受损皮肤接触的非侵入性器械均被分类为II类。具有其他任何预期作用机制或适应症(例如促进愈合、缓解疼痛以及提供湿润伤口愈合环境)并与受损皮肤接触的非侵入性器械,属于II类。
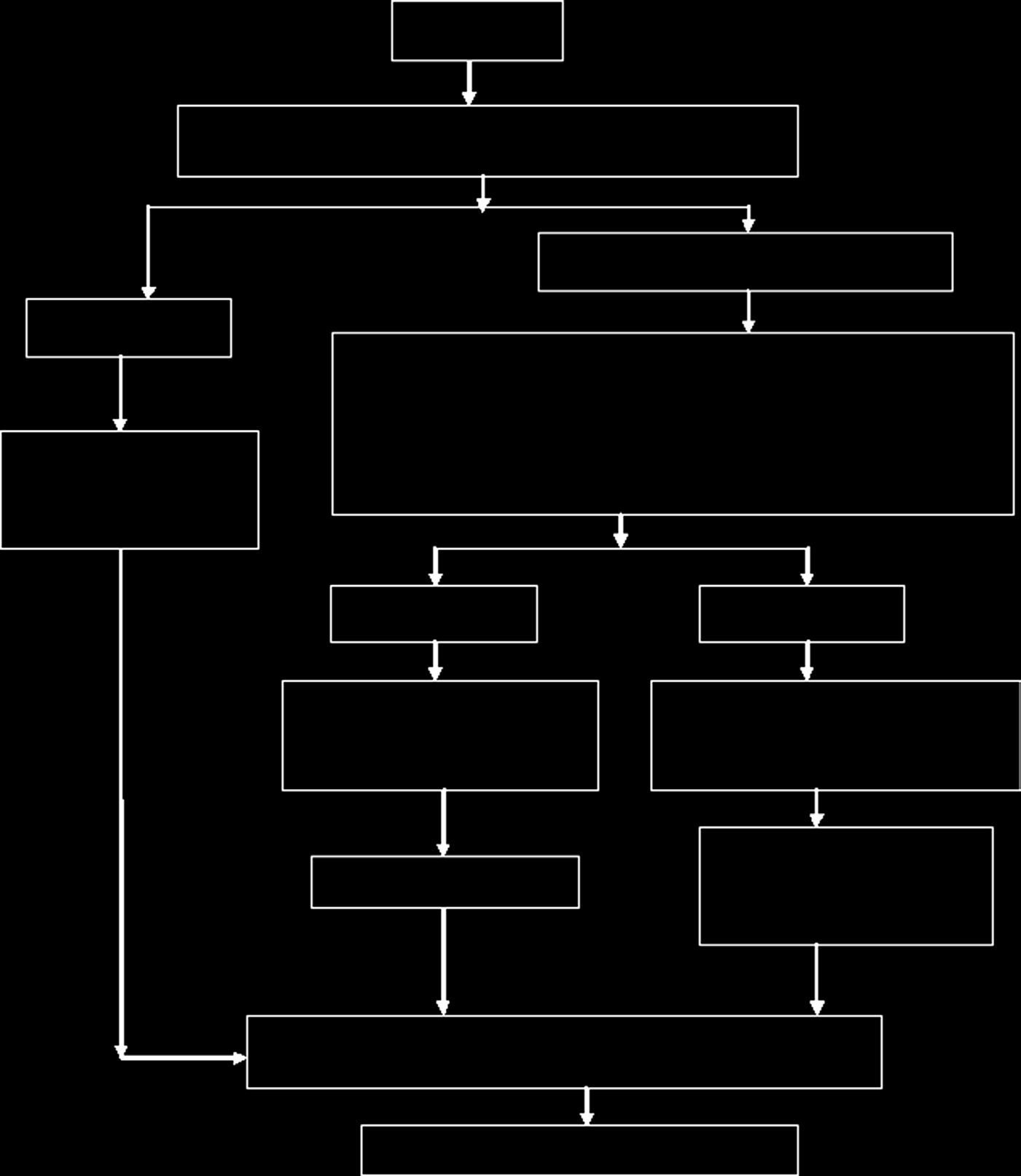
除上述类别外,治疗产品分类委员会还对组合产品进行了分类。根据组合产品的主要作用机制,将采用《医疗器械法规》或《食品和药品法规》对其进行评估。加拿大医疗器械审批流程的一般程序见 图15.14。
15.7 澳大利亚的医疗器械法规
澳大利亚医疗器械市场是全球增长缓慢的市场之一。该市场在2016年的估值为40亿美元,低于2014年的50亿美元。其主要原因是澳大利亚使用的医疗器械中有80%依赖进口[43]。在澳大利亚,治疗用品管理局(TGA)根据1989年《治疗用品法案》和相关法规规定的标准来监管医疗器械的供应Therapeutic Goods Act以及相关法规[44]。这些法规通过全球协调工作组采用的全球协调方法与欧盟法律保持一致[45]。
15.7.1 医疗器械分类
医疗器械根据器械的风险等级和预期用途分为五个等级(表15.10),依据为《2002年治疗商品(医疗器械)法规》第3.2条法规和附表2,以及《1989年治疗商品法》第41BD条。
病床、听诊器、绉纱绷带和轮椅等低风险器械被归类为I类器械。这些器械可通过在线申请在澳大利亚进行注册。I类医疗器械根据其使用方式进一步分为两类,即无菌I类或I类测量器械。无菌I类或I类测量器械的上市销售需要符合性评估证书。该证书必须由治疗用品管理局(TGA)或TGA认可的任何其他监管体系签发[45]。监控设备、手术器械、支架和导管属于IIa类和IIb类器械。
这些器械的上市申请必须附有TGA的符合性评估证书。制造商也可以生产
表15.10 澳大利亚医疗器械分类
| 类别 | 风险等级 | 器械示例 |
|---|---|---|
| I | Low | 手术刀、加压绷带、牙科工具包 |
| IIa | 低中等 | 助听器、麻醉面罩 麻醉 |
| IIb | 中高等 | 避孕套、血袋、输液泵 |
| III | High | 心血管支架、关节假体、心脏瓣膜 |
| 有源植入式医疗器械(AIMD)(有源植入式医疗器械) | High | 起搏器、植入式除颤器 |
在不同监管体系下获得的符合性评估证书,但该体系必须得到治疗用品管理局的认可。
第三类医疗器械(如心脏瓣膜)和有源植入式医疗设备(如起搏器)不仅需要提供符合性评估的证据,还需要对其设计档案进行详细且经认证的评估。
如前所述,任何进口第三类医疗器械的上市申请必须附有由治疗用品管理局(TGA)或TGA认可的其他监管体系出具的符合性评估证明。但对于含有药品、人血或血浆,或含有动物、微生物或重组来源材料的第三类医疗器械,TGA将进行全面的符合性评估。TGA将评估制造商的质量管理体系,该评估可能包括现场审核以及对技术文件的审查。
15.7.2 符合性评估
医疗器械的分类决定了其符合性评估程序。医疗器械制造商需要通过提供与产品以及医疗器械生产过程相关的适当证据,以满足治疗产品立法法案的符合性评估要求。一旦制造商提供了足够的文件以满足治疗产品立法法案的要求(即符合性评估证据),将向其颁发符合性评估证书。这证明制造商已建立适当的体系来生产产品。符合性评估证书是医疗器械的符合性声明,该证书同时也作为将器械列入澳大利亚治疗产品注册清单的申请。
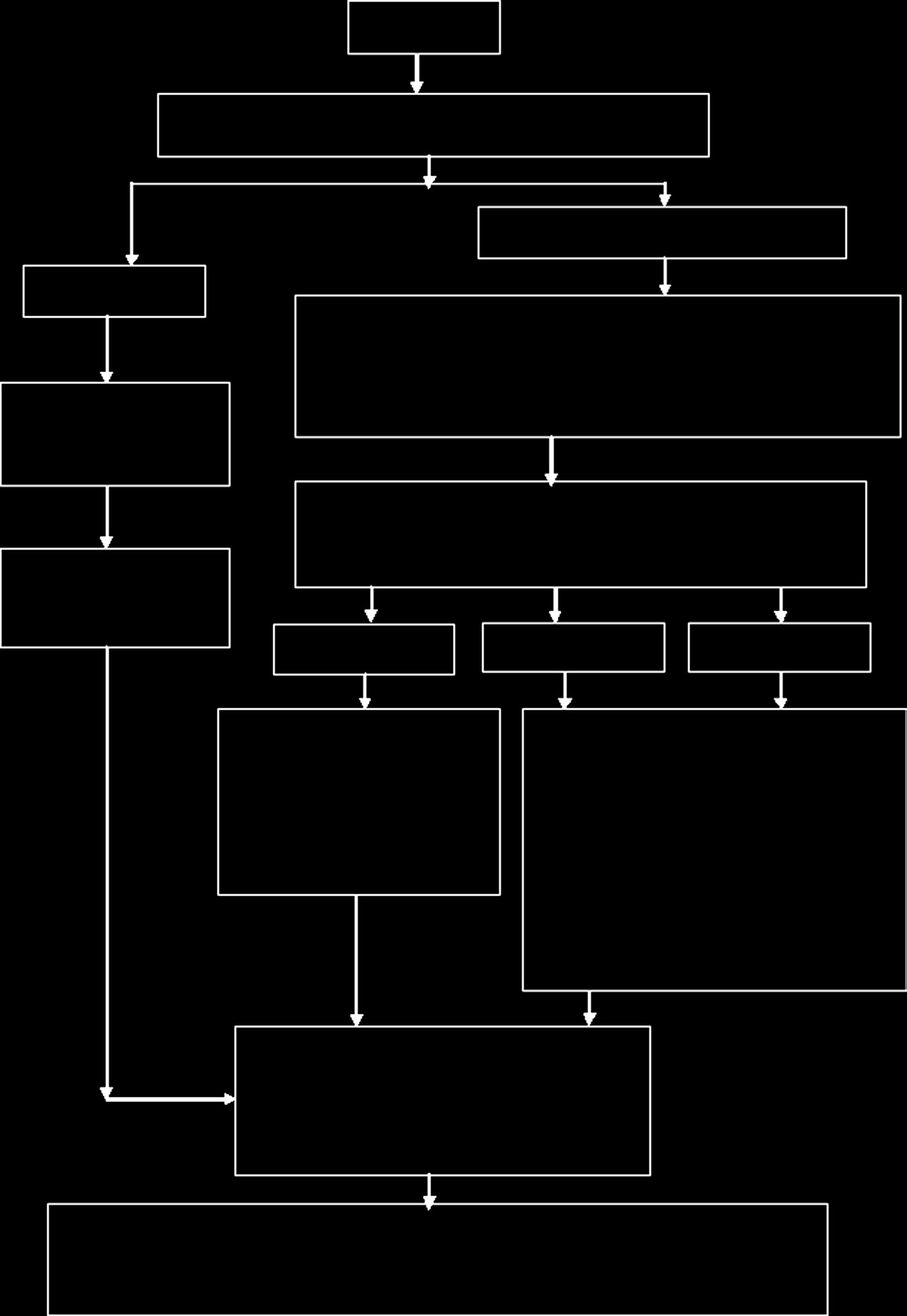
制造商可以选择所需的符合性评估程序,以确保器械经过充分评估,符合该类器械的特定要求。较高分类的器械需要比低类别器械接受更严格的符合性评估。详细的符合性评估程序见图15。15[46]。
15.7.3 基本原则
治疗用品管理局(TGA)的《基本原则》规定了与医疗器械安全性和性能特征相关的要求。《基本原则》要求以下内容:
- 医疗器械的使用不得损害健康和安全。
- 医疗器械的设计和构造必须符合安全原则。
- 医疗器械适用于其预期用途。
- 已确保用户的长期安全性。
- 医疗器械的益处大于任何副作用。
根据医疗器械的性质、设计、功能和制造目的,某些特定的基本原则可能适用或不适用。这些原则包括生物安全性、电气安全、灭菌、生物源性材料、机械和辐射安全以及标签要求等方面的要求。标准医疗器械的制造商将选择适当的标准作为符合基本原则的手段。这些监管标准的示例如下:
- ISO 14971-医疗器械风险管理的应用
- ISO 13485-质量管理体系-监管目的要求
- ISO 11137-医疗保健产品的灭菌-辐射,第1 3部分-制造商应识别相关标准,并记录测试以在器械的技术文件中证明符合性。
澳大利亚医疗器械监管系统中有许多关键参与者,如表15.11所示。
表15.11 澳大利亚监管体系的主要参与者
| 人员 | Role 职责 |
|---|---|
| 制造商 |
执行设计、生产、包装‐ 翻新和标签 该产品。(非个人)
• 确定分类 用途,预期用途和 全球医疗器械命名法(全球医疗器械命名系统) 确保符合合规性 基本原则 • 选择合适的符合性 评估程序 • 符合性声明 |
| 申请人 |
进口、出口或制造 澳大利亚的医疗器械(否 个人)
• 向TGA提供来自制造商的信息 • 提交符合性 评估详细信息 • 将医疗器械纳入ARTG • 向TGA提供样品并允许治疗用品管理局检查并向治疗用品管理局报告不良事件 至治疗用品管理局 |
| 治疗商品管理局(TGA) | 监管治疗的政府机构 治疗性药品和医疗器械 |
| 代理人 | 代表赞助商行事的人员 或制造商包含medi‐ 澳大利亚器械登记册中的器械 |
| TG(澳大利亚治疗商品登记册) |
15.7.4 澳大利亚关于伤口敷料的法规
澳大利亚医疗器械监管指南将上述五类伤口敷料进行分类。提供了不同类型的伤口敷料示例,以便更好地理解 [47]。
15.7.4.1 I类伤口敷料
- 粘性敷料条 – 非无菌、无菌,根据分类规则2.4.3(c);用于扭伤的压缩绷带 – 根据规则2.4(3)(b)
- 吸收垫、岛状敷料、药棉、伤口条带和纱布敷料 – 一种非侵入性器械,用作机械屏障或用于压迫或渗出液吸收,根据规则2.4.3(c)
- 鼻出血敷料 – 根据规则3.1(2)(b)(ii),用于口腔至咽部、耳道至鼓膜或鼻腔的短期使用的侵入性设备
15.7.4.2 IIa类伤口敷料
- 聚合物薄膜敷料、水凝胶敷料和非药物浸渍纱布敷料 – 用于接触受损皮肤的非侵入性器械(包括主要用途为管理伤口微环境的器械)– 根据分类规则2.4(1)
15.7.4.3 IIb类伤口敷料
用于慢性广泛溃疡性伤口、严重烧伤、严重压疮伤口的敷料,或提供临时性皮肤替代物的敷料。该非侵入性器械适用于真皮层已受损且只能通过二期愈合方式愈合的伤口,根据分类规则2.4(4)进行分类 一种用于深层伤口和溃疡的伤口敷料,这些伤口已穿透含有藻酸盐的真皮以吸收渗出物——包含非微生物来源的藻酸盐。通过二期愈合,在分类规则2.4(4)下进行分类
15.7.4.4 III类伤口敷料
含抗菌剂的敷料。——根据分类规则5.1(2)含有药品
- 一种用于渗入真皮的深层伤口和溃疡的伤口敷料,含有用于吸收渗出物的藻酸盐——根据规则5.5(1)(a),包含微生物来源的藻酸盐
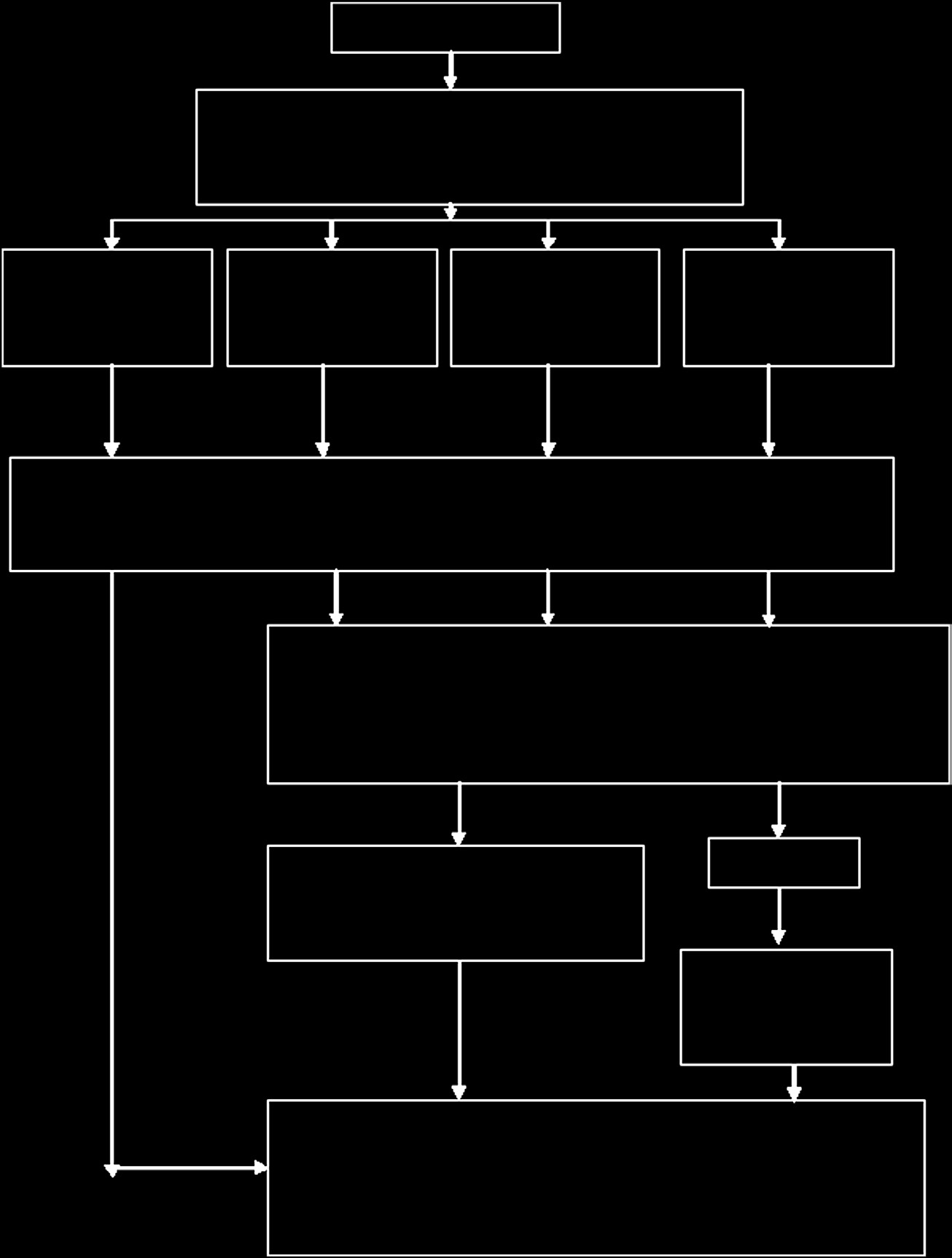
- 一种包含生物源性材料的伤口敷料,例如胶原蛋白、透明质酸钠、硫酸软骨素、表达人胶原蛋白基因的重组植物,依据分类规则5.5.1(a)。医疗器械审批流程如图15.16所示。
15.8 欧盟医疗器械法规的最新变化
以下医疗器械新法规自4月5日起实施2017[48]。该法规取代了现有的指令,具体包括:
欧洲议会和2017年4月5日理事会关于医疗器械的法规(EU) 2017/745,修订指令 2001/83/EC、法规(EC) No 178/2002和法规(EC) No 1223/2009,并废除理事会指令 90/385/EEC和93/42/EEC。
- 欧洲议会和理事会2017年4月5日关于体外诊断医疗器械并

















 6735
6735

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









