







Parallel-Meta Suite:跨平台可交互的微生物组快速分析套件
Parallel-Meta Suite: Interactive and rapid microbiome data analysis on multiple platforms
DOI:https://doi.org/10.1002/imt2.1
发表日期:2022年3月6日
第一作者: Yuzhu Chen(陈俞竹)1,Jian Li(李坚)1
通讯作者:Xiaoquan Su(苏晓泉)(suxq@qdu.edu.cn)1,2
合作作者: Yufeng Zhang(张玉凤), Mingqian Zhang(张明乾), Zheng Sun
(孙政), Gongchao Jing(荆功超), Shi Huang(黄适)
主要单位:
1青岛大学(College of Computer Science and Technology, Qingdao University, Qingdao, Shandong, China)
2中国科学院青岛生物能源与生物过程研究所(Single‐Cell Center, Qingdao Institute of BioEnergy and Bioprocess Technology, Chinese Academy of Sciences, Qingdao, Shandong, China)
图文摘要
Parallel-Meta Suite(PMS)是一个易用的软件包,可以在多个平台上进行快速、全面的微生物组数据分析
PMS涵盖了广泛的数据预处理和统计方法,并提供最新的可视化结果
PMS的整个流程通过并行计算方案进行了优化,可以快速处理数千个微生物组数据
视频解读
Bilibili:https://www.bilibili.com/video/BV12F411s7uM/
Youtube:https://youtu.be/bSdrUSpzNDg
中文翻译、PPT、中/英文视频解读等扩展资料下载,请访问期刊官网:http://www.imeta.science/
摘要
测序通量的提高和测序成本的降低,极大地方便了微生物组研究实验的开展,进而产生了浩如烟海的组测序数据,这些数据中蕴藏着微生物与其环境表型(如宿主健康或生态系统状态)之间的关联。想要破译隐藏在微生物组数据下的生物信息,出色而又可靠的软件工具是不可或缺的。然而现在的大多数的软件,其可用性方面的缺陷为非计算机专业的用户设置了难以逾越的鸿沟。与此同时,计算通量已经成为了许多分析平台处理大规模数据集的一个重要瓶颈。本研究开发了Parallel-Meta Suite(PMS),一个用于快速和全面的微生物组数据分析、可视化和注释的可交互软件套件。PMS采用了最先进的算法,涵盖序列微生物组数据物种与功能解析、统计分析、可视化等一系列流程,并具有友好的图形界面,可以满足各种用户的分析需求。为了适应快速增长的计算能力需求,PMS的整个分析流程都使用并行计算策略进行了优化,具备快速处理上万的样本的能力。此外,PMS还具有多操作系统兼容、简易安装与全自动运行等特性。
引言
想要解读隐藏在微生物组大数据下的生物模式,优秀的生物信息工具是至关重要的。通过这些生物模式,我们可以解释微生物群落与其周围环境(如环境条件或人类健康状况)之间的关联。在过去的十年中,微生物组领域生物信息工具的功能,已经从基本的分类注释扩展到下游的多样性分析和生物标志物的选择,极大地拓展了微生物组数据挖掘的用途。然而,像QIIME或Parallel-Meta这样功能高度集成的工具包,其复杂的命令行式操作给非计算机专业人员的操作,甚至是入门带来了障碍。另一方面,在过去的几年中,测序成本已经大大降低,这促进了不同环境下超大规模微生物的调查与研究,如Earth Microbiome Project或American Gut Project等,同时也提高了对数据处理的计算通量和效率的要求。
面对这样的现状,许多开发者也给出了自己的解决方案,其中一种解决方案是提供基于图形的用户界面(GUI)来提高可用性,例如q2studio。然而,这种图形界面在安装和运行的过程中需要特定的操作系统环境和许多依赖库,这就导致在一些情况下(比如远程登陆服务器和大数据处理)该图形界面无法使用。另一种解决方案是带有图形界面的在线网络服务,例如Galaxy或gcMeta。但是,难以避免的网络延迟和在线计算资源的共享限制了用户分析数据的规模,在进行大量的微生物组测序数据分析时尤为突出。此外,在开放的在线平台上分析未公布的私有数据集时,数据隐私和安全问题也同样令人担忧。
为了应对这些挑战,我们在此开发了Parallel-Meta Suite(PMS),一个用于快速和全面的微生物组分析的软件套件。PMS基于成熟的标志物基因分析协议和工作流程,并进行了重新设计和重大改进,其特点包括(但不限于)提供友好的图形界面提高了多平台下各种用户的可用性、使用全并行计算方案优化分析性能等。此外,为了解决许多生物信息学工具安装困难的问题,如软件包依赖性、系统设置和源代码编译,PMS还集成了自动安装程序,帮助用户轻松安装和配置软件。PMS软件的最新版本已经在GitHub(https://github.com/qdu-bioinfo/parallel-meta-suite)和Gitee(https://gitee.com/qdu-bioinfo/parallel-meta-suite)发布,软件包中还提供了测试用的演示数据集。
方法
图1说明了PMS的分析工作流程。PMS可以接受宏基因组的鸟枪序列或扩增子序列作为原始输入。对于鸟枪法测序序列,利用隐式马尔可夫模型识别和提取标记基因片段(如16S rRNA或18S rRNA基因)。对于扩增子序列,PMS对标记基因进行ASV降噪和去嵌合体,以降低测序错误的干扰(这一步骤对于鸟枪法测序序列默认设置是关闭,也可由用户自行开启)。然后,通过内置的Vsearch将序列与参考数据库进行比对,进行从界级到物种级的剖析和分类学注释。每个分类级别上群落成员的相对丰度也使用标记基因拷贝数进行校正。之后,使用PICRUSt2算法预测功能信息的KEGG Orthology(KO)基因家族,并通过KEGG BRITE层次结构对代谢途径进行注释。PMS还通过NSTI(Nearest Sequenced Taxonomy Index)值来衡量功能的预测准确性,由OTU和它们在系统发育结构中最近的单独测序的亲属之间的距离之和计算出来。
图1 PMS的整个分析流程和可视化的工作流程
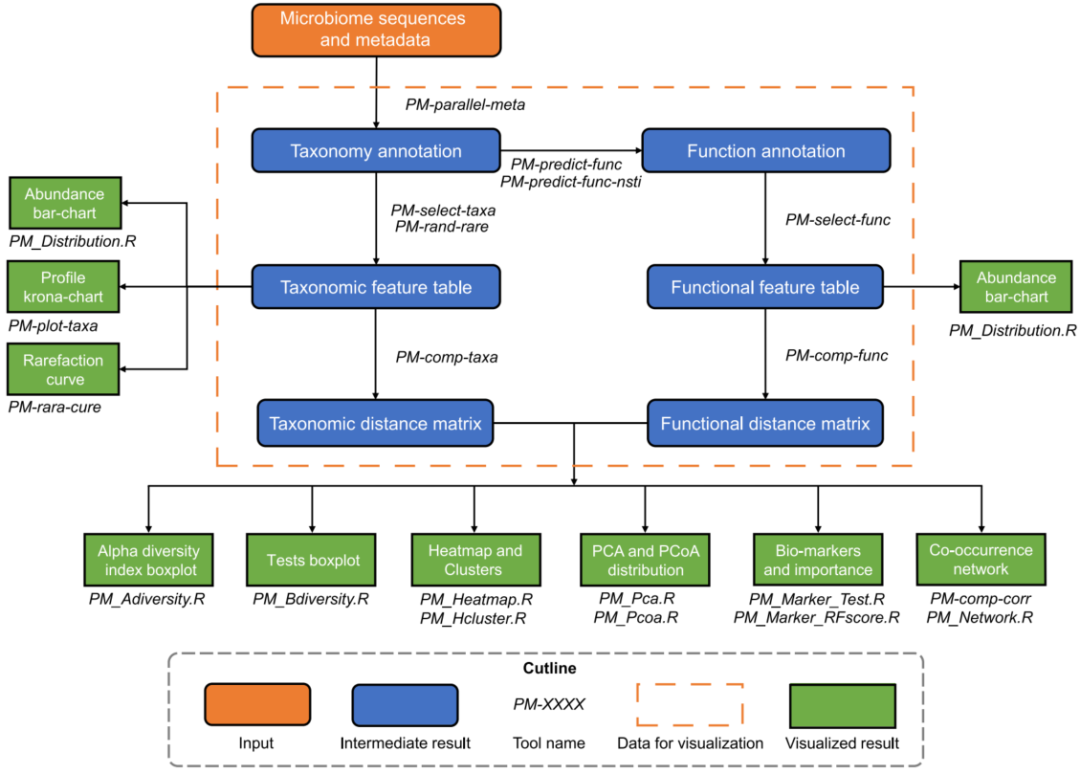
微生物组的物种信息通过Krona和条形图进行可视化。然后,在用户选择的特定分类学或路径级别上进行微生物多样性分析、生物标记物选择和共现网络构建。α多样性分析计算每个样品的香农、辛普森和Chao1指数。对于离散的元数据(如类型、状态、性别等),α多样性指数进行Wilcoxon或Kruskal秩和检验,对于连续变量(如年龄、BMI、PH值等)进行回归分析。β多样性通过加权/非加权Meta-Storms算法(针对物种分类)或Hierarchical Meta-Storms(针对功能)计算所有样本之间距离矩阵,并通过热图进行可视化。之后,通过PCoA(主坐标分析)和PCA(主成分分析)图展示β-多样性模式,对离散元数据进行PERMANOVA和ANOSIM检验,对连续变量和距离值进行回归分析。在生物标志物分析中,PMS使用Wilcoxon或Kruskal秩和检验,选择出在不同组别(离散数据变量)间具有显著差异的微生物或基因单元作为候选标记物,然后通过随机森林的重要性进行排序。与连续变量密切相关的微生物组特征也通过回归分析被挑选出来作为生物标志物。在共现网络中,网络节点是群落特征(例如,一个微生物分类单元),网络的边代表节点间的Spearman相关性,然后计算网络密度、直径、半径和集中度来量化网络属性。
结果
PMS的关键特性
PMS为数据分析的参数配置和详细结果展示提供了一组用户友好的GUI(图2)。用户可以通过一组示例数据和参数配置页轻松上手PMS,该页面图形化的参数展示也降低了用户进阶的学习曲线。该GUI选择网页作为可视化功能的载体,能够兼容不同的使用场景(如本地系统或远程登录服务器)以及多种操作系统(如Linux、Mac或Windows 10)。作为一个高度集成的全自动分析工具,PMS使用了各种最先进的算法和分析策略,包括高级序列处理(如宏基因组标记基因提取,序列ASV(扩增子序列变异)降噪等),16S预测功能谱,α和β多样性计算、多变量统计分析,生物标记物选择和评估,以及共发生网络分析等。标志物基因的参考序列也通过GreenGenes、SILVA、Oral-core、SILVA-18S和ITS数据库进行了更新和扩展,包含全长的16S rRNA、18S rRNA和ITS序列。最后,PMS通过全局的并行化处理和性能调优,使得在一个计算节点上仅耗时40余小时即可完成对14,000例样本的全部分析。
图2 PMS的GUI可视化界面
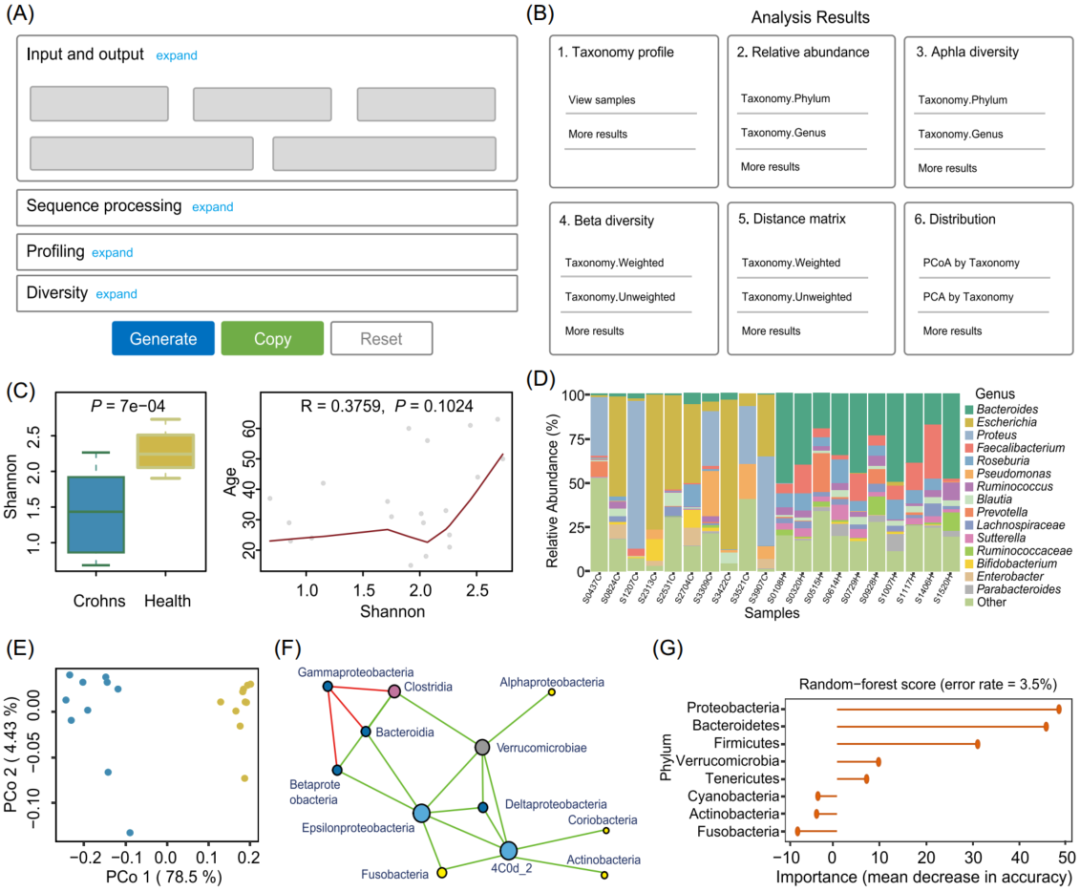
(A) 可交互的配置向导;
(B) 结果导览页;
(C) α多样性计算及其与关键表型的关系;
(D) 样品的相对丰度表;
(E) 基于PCoA (Principal Coordinate Analysis)的β多样性;
(F) 共现网络分析;
(G) 通过随机森林的监督机器学习算法产生的内部重要性分数来选择生物标记。
并行计算和GUI的实现
PMS软件的GUI由两个部分组成,一是交互式的分析参数“配置向导”(图2A),二是可视化的“结果导览”(图2B)。配置向导内置于软件套件之内,其中所有的参数已经按照分析流程进行了分类和整理,以一种易于理解的形式展示出来。在初始状态下,所有的参数已设为默认值,只需填入必要的基本参数(如输入/输出类型和路径)就可以进行分析。配置向导也提供调整高级选项,以进一步对剖析、多样性分析和统计这些步骤进行定制。最后,根据用户的设置,该配置向导可以生成相应的可执行命令。整个分析流程完成后,会在输出目录中自动创建结果导览。该页面会将所有分析结果分类,并通过精心设计的方案和颜色的图来可视化每个结果(图2C-G),为微生物组模式提供直接和清晰的解释。这种图形用户界面对不熟悉命令行界面或复杂参数的非专业用户非常有帮助,也能让他们更好、更清楚地了解分析的工作流程和结果。此外,由于配置向导和结果导览页可以通过任何网络浏览器方便地访问,PMS GUI与包括Linux、Mac和Windows在内的多种操作系统平台高度兼容。
PMS的整体计算框架主要由C++开发,该语言与脚本语言相比,拥有更快的运行速度和更高效的内存使用方式。利用从GUI配置向导中解析出的参数,该框架调用并管理工作流程中的分析步骤。总的来说,我们从两个方面对整个分析流程进行了并行计算方案的优化。(1)对于分类鉴定、丰度估计、功能预测和距离矩阵等有关的计算步骤由C/C++编写,直接由OpenMP库实现并行化。(2)对于α和β多样性和统计检验、生物标记物选择和绘图有关的统计步骤由CRAN-R(https://www.R-project.org)编写,每个R脚本都被PMS计算框架分配到一个独立线程,所有线程可以同时启动以实现并行计算。为了充分利用硬件计算资源,可用线程的数量被默认设定为CPU核心数并动态调整,也可以由用户手动控制。
不同场景下的使用
本小节中,我们将展示PMS在不同计算平台和环境下的三个典型场景(图3)的使用情况和经验。
图3 PMS在不同场景和平台的三种典型使用方式
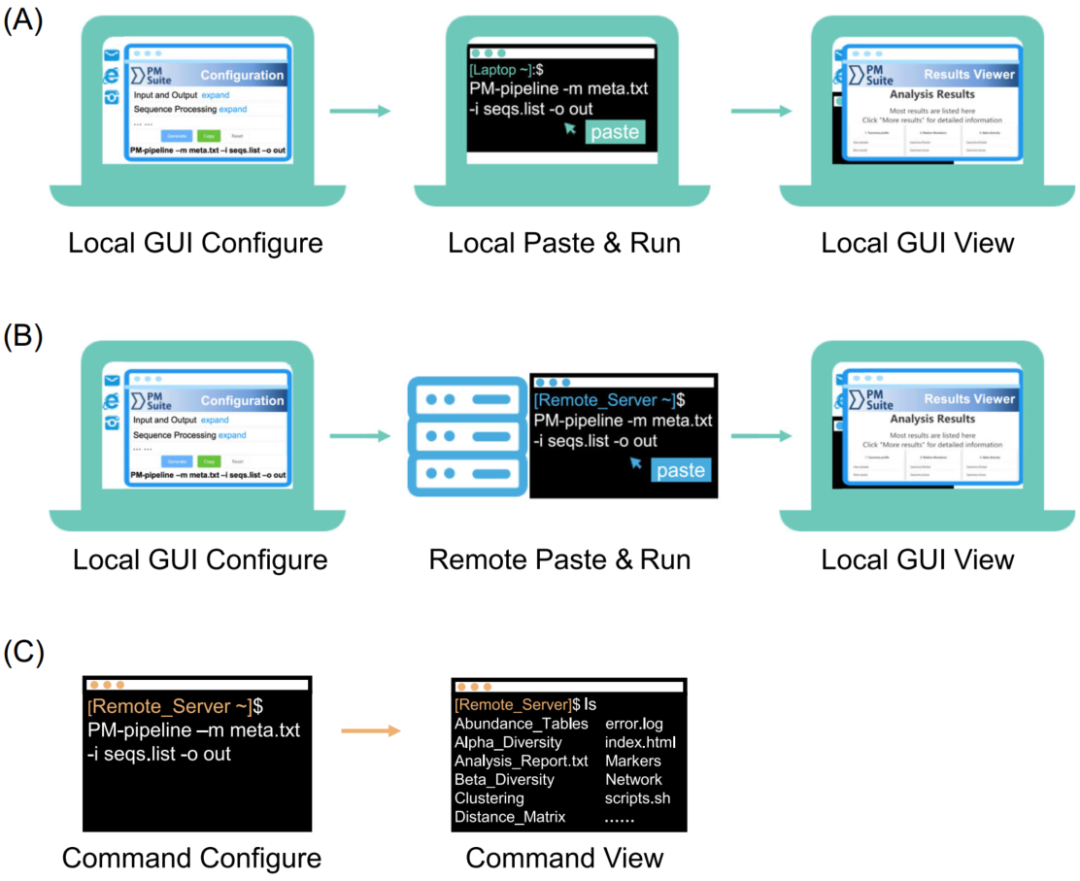
(A) 在本机使用GUI进行参数配置,并在本机进行运算分析;
(B) 在本机使用GUI进行参数配置,并在远程服务器上进行运算分析;
(C) 使用命令行进行参数配置 (本地或远程服务均可)。
场景一:在本地使用GUI进行参数配置,并在本地进行运算分析
PMS可以在“本地”个人电脑(如笔记本电脑)中安装和执行,以处理少量样品(比如少于200)。基于本地GUI的使用方法(图3A)适用于Linux(安装GUI桌面)、Mac或Windows 10+(需要安装Windows Subsystems for Linux(WSL))操作系统。配置向导可以通过PMS-config文件夹中的“index.html”页面访问。用户可以直接使用默认选项,也可以根据实际需求调整参数。配置完成后,通过点击页面底部的“生成”和“复制”按钮,就会生成一条有效的命令并复制到剪贴板中。然后将这个单行命令粘贴在本地终端,就可以成功运行PMS分析流程,而不需要进行其他操作。在输出目录中,可视化的结果导览页也被命名为“index.html”。所有的原始结果(如相对丰度表、距离矩阵等)也会保留,用于进一步深入的数据挖掘或元分析。此外,在结果文件夹中还提供了分析总结、工作日志和详细的分步工作流程脚本。
场景二:在本机使用GUI进行参数配置,并在远程服务器上进行运算分析
大量样本(比如大于1,000)的处理和运算需要更长的时间和更多的计算资源,我们建议在更强大的服务器上运行PMS的分析流程。通常这样的服务器需要远程登录(例如,通过SSH),并且只提供一个基于命令的终端来操作软件。在这种情况下(图3B),用户应在服务器上安装PMS,在本地计算机下载并打开GUI配置向导(下载软件包中PMS-config文件夹,并用浏览器打开其中的“index.html”文件)以生成命令,并在远程服务器的终端上运行这些命令。结果也可以像场景一那样传输到本地计算机上浏览。因此,整个分析流程可以很容易地配置和执行,而无需大量的数据传输。
场景三:使用命令行进行参数配置
PMS也支持基于命令行的操作。此种方式通常是在没有GUI的条件下,针对有经验的用户(图3C)。为了满足微生物组分析中越来越多的用户特定要求,整个分析流程以在高度灵活的设置下工作,例如,用定制的参数运行每个步骤,或者只执行工作流程中的选定步骤。这可以通过本地或远程的基于命令的终端来实现。命令行界面还提供了教程,描述了详细的用法和分析流程在每个单一步骤中的简要帮助信息。
案例研究及结果
我们采用了两个实例数据集来证明PMS在解码微生物组的能力。这两个数据集都是从以往发表的研究中收集的,以便验证PMS分析结果的准确性和可靠性。
案例1:医院开业前后室内微生物组的变化
数据集1包含894个来自医院开业前后室内环境的16S-扩增子微生物组样本(表1)。我们用所有的默认参数执行了PMS分析流程。从结果中我们可以观察到,医院开放后,α多样性的香农指数下降(图4A;Wilcoxon检验p值<0.01),整体群落的β多样性明显转变(图4B;加权Meta-Storms距离,PERMANOVA检验p值<0.01),均已被Lax等人验证过。预测功能多样性遵循与分类学类似的趋势。两个时间点之间的这种微生物动态也可以通过相对丰度的变化来说明(图4C)。使用统计测试和机器学习分析方法,PMS还确定了有助于区分医院表面从开业前到开业后状态的这种生态变化的最重要的微生物,如葡萄球菌、莱茵海拉菌和莫德斯特菌。这个机器学习模型在区分室内样本(图4D)的属级状态方面达到了95.91%的准确率(误差率=4.09%)。
表1 测试数据集的详细信息
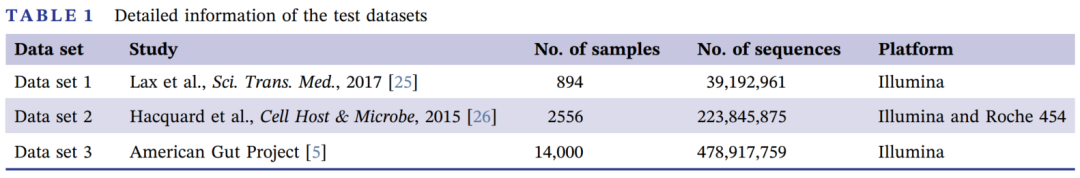
图4 医院开业前后室内微生物组的变化
(A) 医院开业后,α多样性的香农指数下降,Wilcoxon测试P值<0.01(P值<0.05表示差异显著);
(B) 根据加权的Meta-Storms距离,开院前和开院后状态下的整体β多样性有显著差异,PERMANOVA检验P值<0.01(P值<0.05表示有显著差异);
(C) 两个时间点之间属水平的相对丰度的动态变化;
(D) 五种细菌属被选为可以区分两个时间点的生物标志物。X轴是随机森林模型产生的重要性得分(准确性的平均下降),该模型评估了每个生物标志物对区分不同医院状态的重要性。
案例2:来自多个栖息地的微生物组的元分析
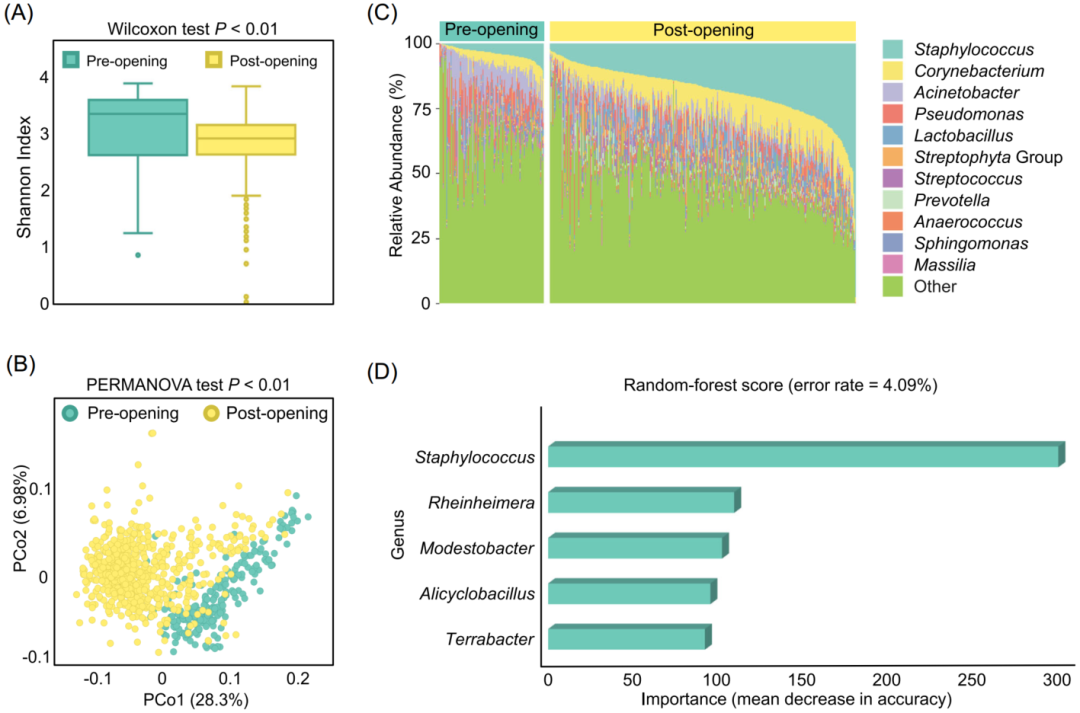
数据集2包括2,556个宿主相关的微生物组(表1),取自不同的宿主物种和研究,我们从中进行了元分析,以系统地研究微生物在不同环境栖息地的分布。由于16S rRNA基因扩增子序列是由不同的平台(即Illumina和Roche 454)产生的,不适用于ASV降噪,其他选项被保留为默认值。图5A和图5B的结果显示,PMS揭示了宿主来源或栖息地类型之间的微生物组的不同α和β多样性。这主要是由于哺乳动物肠道和植物根部之间丰富的类群很少重叠,而鱼类肠道和植物根部群落有共同的微生物成员,例如,蛋白质细菌、蓝细菌和放线菌的优势菌群(图5C),与之前的研究Hacquard, et al., Cell Host & Microbe 2015高度吻合。同样有趣的是,功能上的α和β多样性产生了与分类学类似的结果,然而,所有样品在KEGG BRITE 2层级的一些代谢途径却较为一致,如蛋白质家族遗传信息处理、信号传递和细胞处理、碳水化合物代谢、氨基酸代谢和能量代谢。
图5 对来自多个栖息地的微生物组进行元分析
(A) 宿主类型之间的α多样性香农指数显著不同。Kruskal检验P值<0.01(P值<0.05表示有显著差异);
(B) 在基于PCoA模式的加权Meta-Storms距离中,样本按栖息地进行分组。PERMANOVA测试P值<0.01(P值<0.05表示有显著差异);
(C) 在不同的栖息地类型中,丰富的群落成员有所不同。
并行计算和运行速度
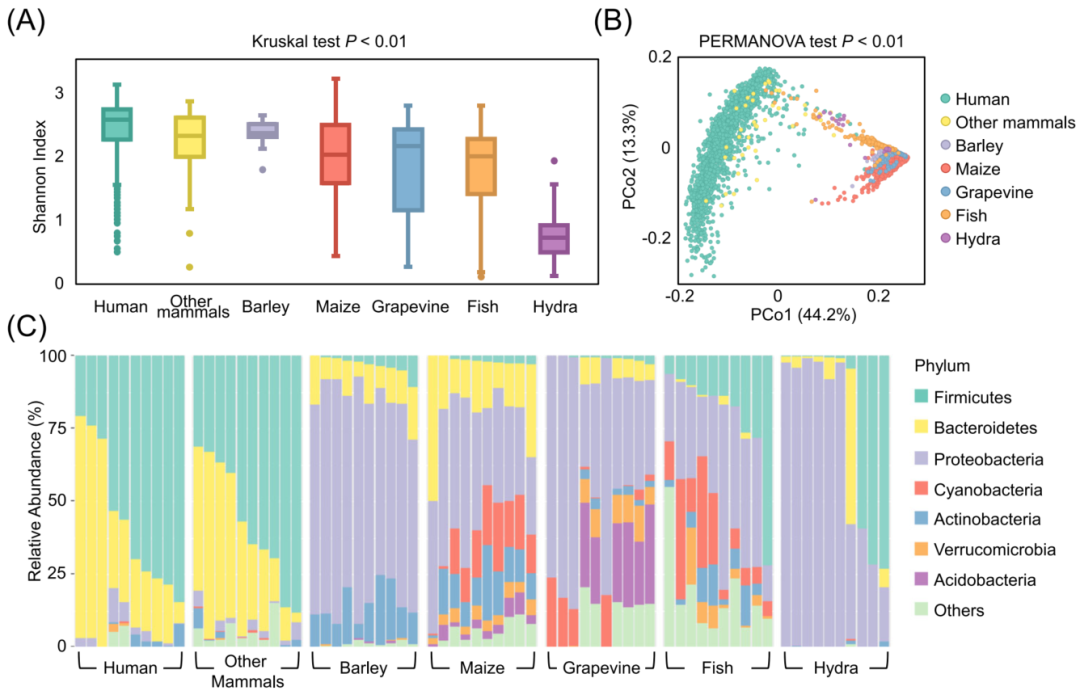
我们利用三个数据集(表1)进一步评估了PMS在并行计算速度和效率方面的表现。对于数据集1和数据集2,我们分别设置了不同数量的CPU线程(1,10,20,40和80),重复整个工作流程并比较运行时间以测试并行计算的效率。数据集1和3是由Illumina平台测序的,适用于基于ASV的分析。数据集2包含Illumina和Roche 454的序列,所以ASV被设置为关闭。其他参数保持为默认配置。所有的速度测试都是在一个支持80个线程(40个物理CPU核)的单节点机架服务器上进行的。
通过动态线程调度和并行计算的负载均衡的优化,PMS能够处理成数以万计的微生物组数据,例如,超过2,500个样本的数据集2的整个工作流程可以在392分钟内完成,甚至数据集3的14,000个样本可以在43小时内完成。从图6的结果中,我们观察到运行时间的减少与线程数呈线性关系,表明并行化和子任务调度策略的计算效率很高。此外,加速比与输入样本的来源或序列类型无关。这种加速表明,PMS可以快速及时地对输入的样本进行分类和功能分析,这对于来自不同技术背景的一万多个样本的深度数据挖掘是至关重要的。
图6 Parallel-Meta Suite在不同规模数据集上的运行时间消耗
讨论
在过去的几年中,微生物组的数据处理方法不断更新并趋于稳定,生物信息工具的关键重点正在从单纯的功能扩展向易用性转变。作为一个不断维护和迭代的软件作品,Parallel-Meta Suite旨在为不同背景、不同水平的用户提供愉快的工作体验,并通过最新的方法或技术提供全面的、迅速的微生物组大规模分析解决方案,有助于利用广泛的数据集形成综合的微生物组知识库,促进跨学科合作。
此外,PMS还通过其高度兼容性促进了深入的数据挖掘。首先,它的数据可视化结果可以提供对与关键表型相关的微生物多样性模式的清晰理解,并为下游分析或更大规模的研究产生某些关键假设。另一方面,所有的原始数据都以标准或常用的格式存储,用于大数据挖掘的下游过程。例如,具有不同微生物特征集(如分类学或功能途径)的相对丰度表也适合其他微生物组分析工具或机器学习工具。这样的微生物组分析结果可以直接被我们之前开发的工具,如微生物组搜索引擎或Meta-Apo无缝使用,这大大促进了该领域的数据驱动科学。
代码和数据可用性
该软件包现已在GitHub(https://github.com/qdu-bioinfo/parallel-meta-suite)和Gitee(https://gitee.com/qdu-bioinfo/parallel-meta-suite)发布,其中集成了一个安装程序以实现全自动安装。文中所用的所有数据集也都上传到了线上的仓库中。在每个数据集包中,“seqs”文件夹包含每个样品的解复用FASTA格式的序列文件,“seqs.list”文件记录了这些样品序列的路径,“meta.txt”文件则包含每个样品的meta信息。所有的补充材料(文本、图、表、中文翻译版本或视频)也可从线上获取。
责编:马腾飞 南京农业大学
审核:iMeta期刊编辑部
作者简介

陈俞竹,青岛大学软件工程学术硕士,2019年公派至瑞典布莱津理工大学交换学习。目前研究方向为微生物组大数据分析与挖掘,相关学术成果已发表于iMeta、Computational and Structural Biotechnology Journal等期刊。

李坚,青岛大学电子信息专业硕士。前中兴通讯工程师,后考入青岛大学攻读硕士学位。目前研究的主要课题为微生物组分析工具。
通讯作者简介

苏晓泉,青岛大学教授,博士生导师。研究方向为生物信息学与大数据科学,已在iMeta、mBio、mSystems、Bioinformatics等期刊发表学术论文40余篇,主持国家自然科学基金项目、国家重点研发子课题、山东省自然基金重大基础项目、中科院重点部署项目子课题等,相关成果获得8项软件著作权。
引文
Yuzhu Chen, Jian Li, Yufeng Zhang, Mingqian Zhang, Zheng Sun, Gongchao Jing, Shi Huang, Xiaoquan Su. 2022. Parallel-Meta Suite: Interactive and rapid microbiome data analysis on multiple platforms. iMeta 1: e1. https://doi.org/10.1002/imt2.1
iMeta—微生物组/生物信息高起点期刊

联系方式:
主页:http://www.imeta.science
出版社:https://onlinelibrary.wiley.com/journal/2770596x
投稿:https://mc.manuscriptcentral.com/imeta
邮箱:office@imeta.science
微信公众号:iMeta
iMeta文章中文翻译+视频解读
iMeta教你绘图
iMeta相关资讯
往期精品(点击图片直达文字对应教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|




























后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集




















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









