






点击蓝字 关注我们
Review:用于蛋白质结构预测的宏基因组定量分析

https://doi.org/10.1002/imt2.9

2022/3/6
● 2022年3月6日,华中科大宁康组在iMeta在线发表题为“How much metagenome data is needed for protein structure prediction: The advantages of targeted approach from the ecological and evolutionary perspectives”的综述型文章。
● 该文章从生态和进化模式角度出发,解码了宏基因组数据与蛋白质结构的复杂关系,并在宏基因组数据中发现了可用于高效补充蛋白质同序列的靶向方法。在有效利用宏基因组数据搜索同源序列并预测蛋白质结构方面具有指导意义。
● 第一作者:杨朋硕
● 通讯作者:宁康
(ningkang@hust.edu.cn)
摘 要
大量工作已经证明,使用宏基因组数据可以补充蛋白质的同源序列来预测其三维结构。然而,尽管一系列工作使用了大量的宏基因组数据,但我们仍然尚未获得很大一部分蛋白质的可靠结构。在本工作中,我们专注于定量的分析宏基因组数据中蕴含的微生物群落生态和进化模式,解码这些模式与蛋白质结构的复杂关系,并研究如何有效地利用这些模式来提高蛋白质结构预测的效率和准确性。首先,我们提出了宏基因组“利用效率”和“边际效应模型”来量化蛋白质家族的同源序列在不同生态位(即生存环境,biome)中的分布模式。其次,在宏基因组数据的搜索策略上,与传统的非靶向方法的盲搜索策略相比,本研究指出了靶向方法的优势:通过搜索来自指定生态位的微生物组数据,能够更有效地补充蛋白质同源序列。最后,我们评估了预测当前 Pfam 数据库中所有蛋白质结构所需的宏基因组数据的下限,并得出目前的宏基因组数据还远未能够达到此目的的结论。总之,在本工作中,我们从生态和进化模式角度出发,解码了宏基因组数据与蛋白质结构的复杂关系,并在宏基因组数据中发现了可用于高效补充蛋白质同序列的靶向方法。本工作在有效利用宏基因组数据搜索同源序列并预测蛋白质结构方面具有指导意义。
关键词:生态学,进化,宏基因组数据,蛋白质 3D 结构建模, 靶向方法
亮 点
● 利用宏基因组补充蛋白质的同源序列对其三维结构预测有益
● 宏基因组利用效率揭示了来自不同生态位数据的蛋白质同源序列数量的差异
● 边际效应模型建立了蛋白质同源序列和宏基因组数据来源生态位之间的关系
● 对于挖掘同源序列,靶向方法优于非靶向方法
● 当前的宏基因组数据不足以支撑预测Pfam 数据库中所有蛋白质结构
Bilibili:https://www.bilibili.com/video/BV1tY4y1H7B1/
Youtube:https://youtu.be/8Qb_6G11mvY
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
引 言
当前大量的工作已经证明了宏基因组序列可以补充蛋白质的同源序列从而准确构建其三维(3D)结构。然而,尽管大量宏基因组数据被用于辅助蛋白结构预测,但仍有相当数量的蛋白质3D结构无法被准确建模。这种现象引出了一个疑问:对于利用微生物组序列辅助预测蛋白结构这一问题,是否存在依赖于宏基因组数据分布的模式;呼应这种分布模式,蛋白质结构的复杂但重要的特性是什么;以及如何更好地利用这些特性来预测蛋白质结构。更重要的是,这背后的原因,很有可能与不同生态位(即生存环境,biome)中微生物群落的生态和进化规律密切相关。
在该研究中,我们专注于解析宏基因组中蛋白质家族的同源序列在不同生态位中的分布差异,并对宏基因组的蛋白质 3D 结构预测方法进行了评估。首先,为了检测不同生态位中,宏基因组的同源序列的差异分布,该研究提出了宏基因组利用效率(utilization efficiency)的概念,将其定义为在宏基因组序列中搜索到的蛋白质同源序列的比例。从生态和进化的角度对不同生态位下的宏基因组利用效率的分析表明,存在生态位特异的蛋白质家族同源序列分布。其次,该研究构建了边际效应模型(marginal effect model),来量化不同生态位中的宏基因组在补充同源序列方面的潜力差异。第三,基于这种同源蛋白的分布差异,靶向方法(targeted approach)可以通过预测和利用能够补充足够同源序列的生态位中的微生物群落,比非靶向方法的盲目搜索更精准和有效地补充其同源序列。基准测试结果表明,与非靶向方法相比,靶向方法可以使用更少的宏基因组序列,并且预测出更精确的蛋白质3D结构。最后,该研究估计了预测当前 Pfam 数据库中所有蛋白质结构所需的宏基因组数据的下限。结果表明,当前的宏基因组数据(大约 1.48E12 条宏基因组序列)不足以对Pfam 数据库中所有蛋白质构建可靠的3D结构(大约需要7.12E12 条宏基因组序列)。但是由于微生物组利用效率更高,所以靶向方法可以使用更少的微生物组序列(大约 4.32E12条)来部分解决这一挑战。
总的来说,我们通过评估不同微生物组数据的利用效率和构建边际效应模型,发掘了宏基因组数据背后复杂的生态和进化规律。利用这些模式和规律,靶向方法可以高效地挖掘同源序列和预测蛋白质3D结构。
蛋白质3D结构预测
一直以来,蛋白质结构如何与蛋白质功能相关联一直是未知的挑战。作为一种计算模拟方法,蛋白质的三维 (3D)结构预测是理解这一问题的关键手段。然而,蛋白质在形成最终3D结构之前,理论上蛋白折叠方式的多样性是天文数字级别的。然而,蛋白质在自然界中会自发折叠,有些在几毫秒内就完成折叠——这种理论和现实的差异被称为 Levinthal 悖论。此外,一些已发表的工具给我们提供了研究数亿种目前缺乏结构的蛋白质的能力。这些未知结构的蛋白质中可能存在具有新颖而有趣功能的蛋白质。这些工具和未知结构蛋白的关系就像望远镜可以让我们更深入地观察未发现的宇宙一样。
蛋白质3D结构的测定通常通过实验观察。X 射线晶体衍射、NMR 光谱和电子显微镜是目前用于鉴定蛋白质结构的常用技术。为了确定最终的蛋白结构,科学家经常综合使用几种不同的方法。然而,由于实验过程通常缓慢而艰巨,因此对于大量未知结构的蛋白质,通常采用计算方法来预测蛋白质的 3D 结构。
不依赖于模板的蛋白质 3D 结构预测
蛋白质 3D 结构通常通过两种方法进行预测:依赖于模板和不依赖于模板的方法。依赖于模板的蛋白质结构预测(也称为同源性或比较建模)利用已有的蛋白结构来模拟蛋白质序列的天然或真实折叠。长期以来,依赖于模板的蛋白质结构预测一直被认为有能够预测出接近天然构象的蛋白模型的巨大潜力。然而,由于依赖于模板的方法强烈依赖于已知蛋白结构,它只能作用于有限数量的蛋白质。
不依赖于模板的方法由大数据驱动,基于同源蛋白质序列和多序列比对 (MSA),无需任何已知模板即可预测蛋白质结构。不依赖于模板方法仅依赖于大量高质量的同源序列来预测准确的蛋白结构。目前,不依赖于模板的预测方法被广泛用于蛋白质 3D 结构预测,包括 Rosetta、I-TASSER和 AlphaFold2等。Rosetta是一个长期更新的用于预测蛋白质结构的软件系统,以其多功能和多样化的应用而闻名。I-TASSER也是一个长期更新的蛋白质结构预测软件系统。借助深度学习方法,I-TASSER 在不依赖于模板的蛋白质结构预测领域表现出色。最近的 CASP14 竞赛中,基于深度学习的AlphaFold2 在92 个未知结构的蛋白质中预测了87 个极高精度结构,优于其他方法。所有这些无模板工具的成就很大程度上依赖于同源序列,这意味着同源序列对于不依赖于模板蛋白质 3D 结构预测至关重要。
总之,不依赖于模板的方法在蛋白质3D结构预测中越来越常见。一方面,深度学习技术使不依赖于模板的方法能够以前所未有的速度和准确性预测蛋白质结构。另一方面,无模板方法通常依赖于蛋白质的同源序列,这些序列本身是丰富多样的。对同源序列的要求已经形成了不依赖于模板的蛋白质 3D 结构预测的一大特点和潜在瓶颈。
不依赖于模板的蛋白质 3D 结构预测的当前问题
任何事物都有两面性,不依赖于模板的蛋白质 3D 结构预测也不例外。一方面,目前的蛋白预测方法,特别是 AlphaFold2,已经能够对 21 个物种的超过 365,198 种蛋白质(平均80.45%的蛋白)进行准确的结构预测。在六个物种中几乎所有蛋白质(超过 99 %)都能被预测。另一方面,许多蛋白质,包括 Pfam 数据库中的蛋白质,都没有已知的 3D 结构,而且这个数字仍在飙升:在 Pfam 发布版本26.0 中,只有 2% 的蛋白质缺乏结构信息,但在 Pfam 发布版本34.0 中,超过 50% 的蛋白质缺乏结构信息(图 1)。这些事实指向了一种表面上矛盾但合理的趋势:在越来越多的蛋白质结构被精确地预测的同时,也出现了越来越多未知结构的蛋白质。这是合理的,因为越来越多的物种被测序,导致越来越多的蛋白质被鉴定。其中许多是新蛋白质,因为缺少足够的同源序列信息而无法快速而精准的建立它们的 3D 结构。面对越来越多的新蛋白质,迫切需要有效地找到其足够的同源序列,用于蛋白质 3D 结构预测。
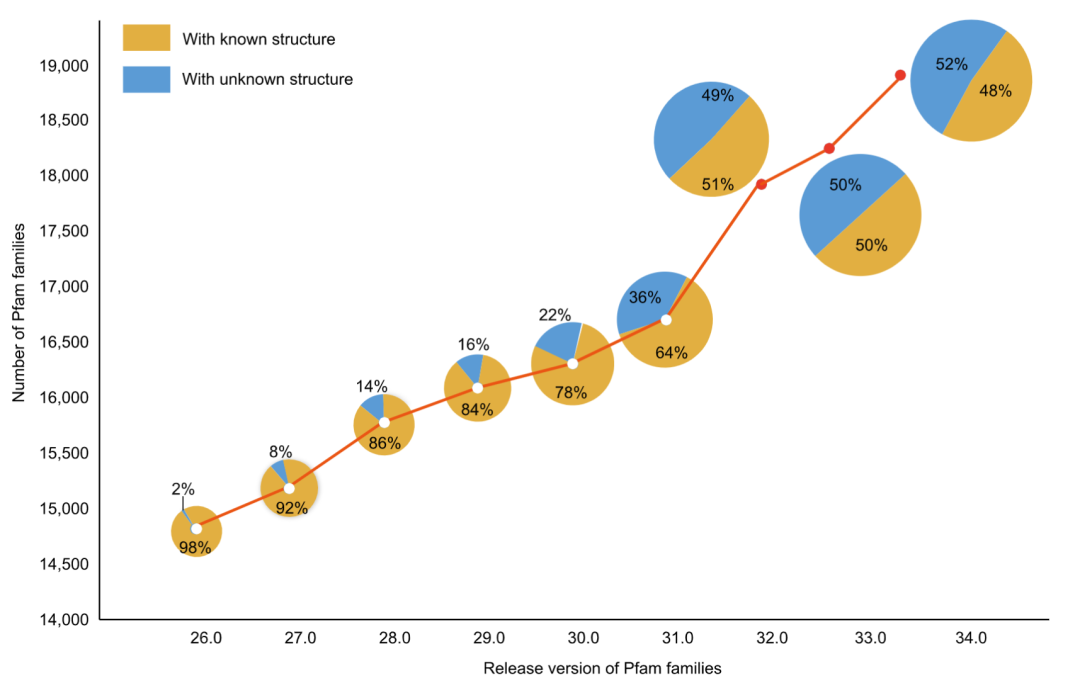
图1. 不同发布版本中的 Pfam 蛋白家族中蛋白质数量和其结构信息的变化趋势
该曲线显示了不同发布版本中Pfam蛋白家族的数量。发布版本对应的饼图反映了已知和未知结构的 Pfam蛋白家族的比例
使用宏基因组序列预测蛋白质 3D 结构
为不依赖于模板的蛋白质 3D 结构预测方法提供同源序列的一种解决方案是,通过使用宏基因组序列来补充其同源序列:作为含有大量功能基因的数据资源库,宏基因组可以为蛋白质提供大量同源序列。结合更加充足的同源信息和先进的不依赖于模板的预测流程,许多未知结构的蛋白质将被预测出准确的结构。
然而,无论使用何种蛋白质结构预测技术,“更多的宏基因组序列可以辅助预测出更多的蛋白质结构”在大多数情况下都是不正确的。在2017年的一项工作中,Baker 等人使用来自不同生态位的宏基因组样本(主要来自肠道生态位)的超过 20 亿条蛋白质,预测了 Pfam 数据库中未知结构的 614 种蛋白质的3D结构。在2019年的一项工作中,虽然仅利用来自海洋宏基因组数据的 9700 万条蛋白质,但 Zhang 等人可以预测 Baker 等人的工作中无法预测的27种蛋白质的结构。在2021年的一项工作中,通过使用来自四个生态位(肠道、湖泊、土壤、发酵罐)微生物群落中的42.5 亿条微生物组蛋白质,Yang 等人可以预测 Pfam 数据库中 1,044 种未知结构的蛋白质结构。所有这些发现表明,宏基因组序列可以补充蛋白质 3D 结构预测的同源序列,并且这种补充具有显著的生态位差异性。
因此,两个很明显的问题值得深入探索:有效利用宏基因组数据进行蛋白质结构预测的手段是什么?蛋白质结构预测需要多少宏基因组数据?对于这两个问题,其核心解决途径只有一个:有效的同源序列补充。研究哪些因素影响了从宏基因组数据补充同源序列的效率,并找到更好地利用宏基因组数据分布特性来辅助预测蛋白质 3D 结构的方法,是非常关键的。为了回答这些关键问题,我们从生态和进化的角度探究了宏基因组数据背后的数据依赖模式(图 2):使用宏基因组数据补充后成功建模的蛋白质作为研究对象,我们探索了相关同源序列的进化模式(同源序列的数量和对应蛋白质功能)和生态模式(来源物种和来源生态位的富集模式)。
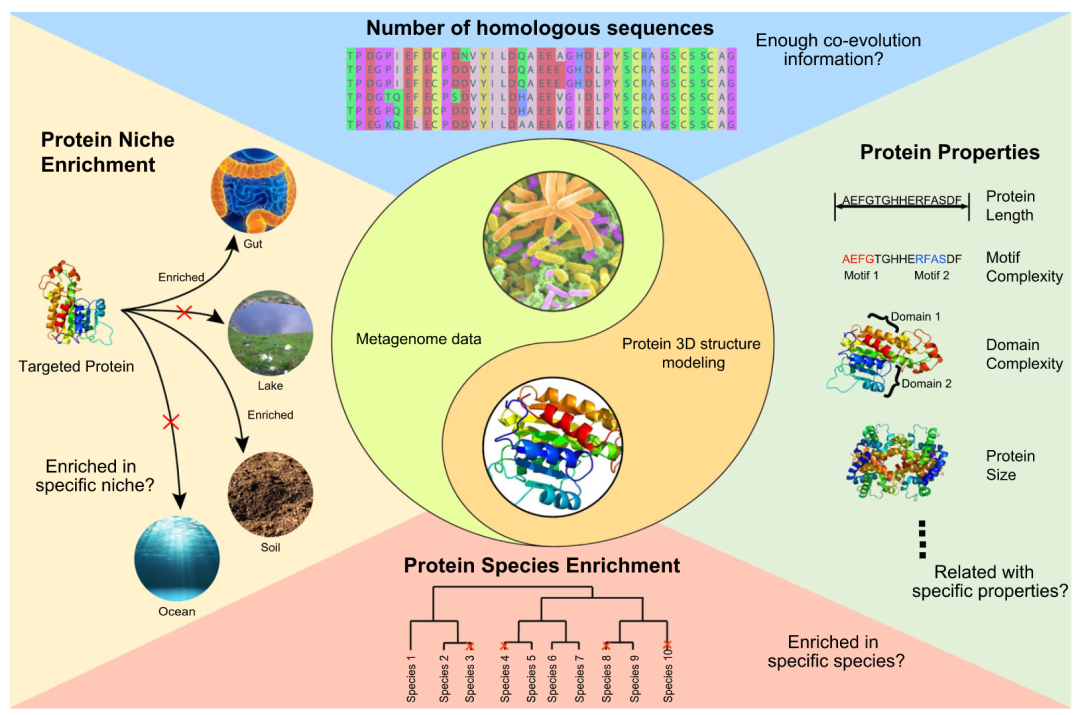
图 2.探究宏基因组数据背后的生态和进化模式
为了探究宏基因组序列和Pfam 数据库中蛋白质之间的相关性,我们研究了相关同源序列的进化模式(同源序列的数量;蛋白质功能)和生态模式(来源物种和来源生态位的富集模式)
计算宏基因组利用效率
随着微生物组数据的爆炸式增长,在宏基因组中搜索蛋白质的同源序列需要巨大的搜索空间和大量的时间,纯粹依靠增加微生物组的数据量,显然是不明智的。因此,提高宏基因组利用效率是成功预测蛋白质3D结构的关键。“宏基因组利用效率(Metagenome utilization efficiency, UE)”被定义为在给定蛋白质的条件下,在特定的微生物数据集中能够作为蛋白质同源序列的比例。显然,更高的宏基因组利用效率值表明微生物序列数据集对相应蛋白质结构预测更有效。如何提高宏基因组利用率的途径也很清楚:预测具有较高“宏基因组利用效率”的微生物序列数据集来限制蛋白质序列的搜索空间。本工作评估了使用来自不同生态位的宏基因组补充同源序列的有效性(图 3)。
首先,对来自不同生态位的宏基因组,搜索所有 Pfam蛋白家族的同源序列后,评估这些微生物序列数据集的同源序列数量(图 3 A),该分析依赖于公开的微生物群落数据,主要来自于4个生态位:肠道、土壤、湖泊、发酵罐,以及 IMG 数据库中的数据。然后,以每十亿宏基因组蛋白质为单位,计算得出能够补充补充同源序列的数量以及能够获得可靠结构的蛋白质的数量(图 3 B)。对于来自四个生态位(土壤、湖泊、发酵罐和肠道)的组合数据集,使用42.5 亿条宏基因组蛋白,对1,044 个 Pfam 家族预测出了高度可靠的模型,占 8,700 个未知结构的 Pfam 蛋白家族的 12.00%,高于之前的研究结果,也高于只使用四个生态位之一的结果。然而,利用组合数据集降低了微生物组数据使用效率。以土壤生态位作为单个生态位的代表,在土壤检测到9.1e+5条Pfam蛋白家族的同源序列,利用效率为6.5e+5(9.1e+5个同源序列/14亿蛋白质)。然而,对于组合数据集,虽然检测到 14.6e+5 条Pfam蛋白家族的同源序列,但宏基因组序列的利用效率仅为 3.4e+5(14.6e+5 条同源蛋白/43 亿同源蛋白),远低于土壤生态位的利用效率。当使用 IMG 数据库(包括来自多个生态位的微生物群落)时,也会得出类似的结果(图 3 B)。以上宏基因组利用效率分析表明,如果我们针对特定蛋白质家族指定其来源生态位,那么单个生态位的蛋白质序列的使用效率会显著高于使用来自不同生态位的组合数据集。
总而言之,宏基因组利用效率极其依赖于生态位的选择。这一现象可以根据基因或蛋白质进化的生态学观点进行解释:在给定生态位的特定环境压力下,一些基因会发生进化,从而使宿主物种能够更好地适应环境。点突变或基因结构变异可能在此过程中出现并积累。因此,我们经常可以在一个生态位中找到特定蛋白质的众多同源序列。进一步来说,为单个蛋白质选择合适的生态位,限制蛋白质序列的搜索空间,将大大提高宏基因组的利用率,并有助于推断其蛋白质结构和功能。
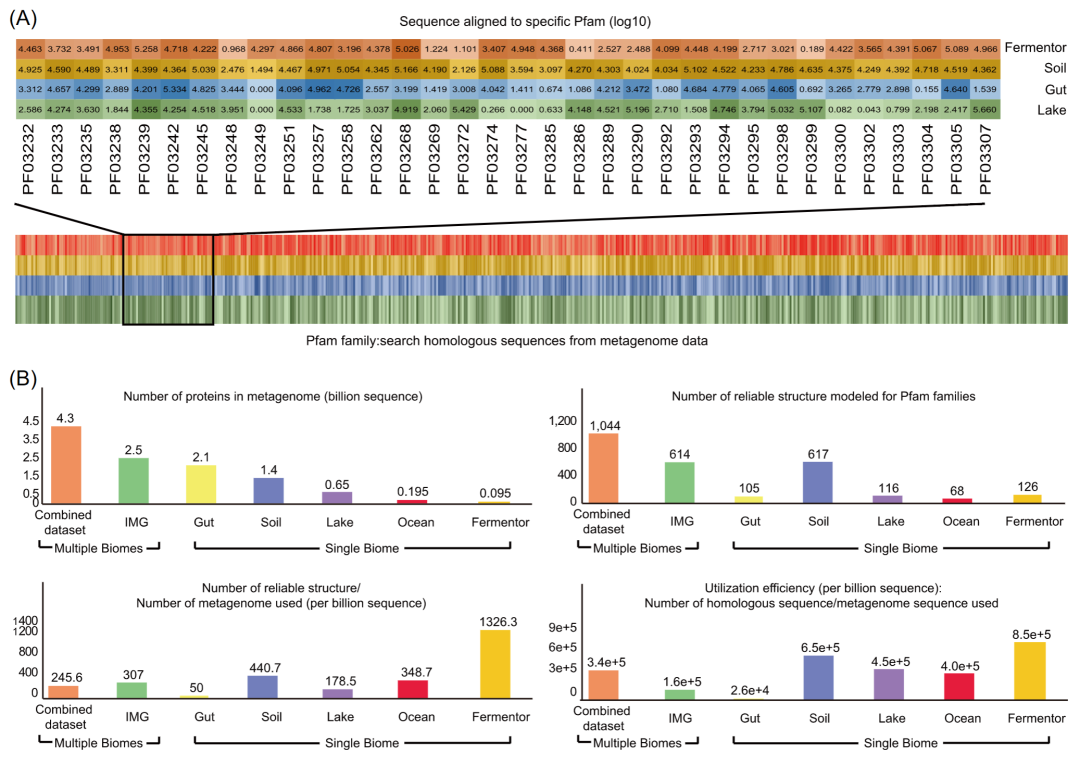
图 3. 宏基因组利用效率评估
A.来自不同生态位的宏基因组数据集。以四个生态位的宏基因组为例,展示与所有 Pfam蛋白家族比对后,其同源序列的分布。不同的颜色表示它们的不同来源生态位,颜色的深浅表示在相应Pfam蛋白家族中比对上的宏基因组序列的数量(颜色越深,序列比对上的越多)。
B. 同源序列比对后,预测的微生物组数据利用效率。在使用宏基因组序列(以十亿为单位)后,计算了比对上的同源序列数和建模的可靠结构数量。为了评价宏基因组利用效率,通过预测出的Pfam蛋白家族数占宏基因组序列数的比例,以及比对上的同源序列数在所有宏基因组序列中的比例。
蛋白质结构预测的边际效应
“边际效应(Marginal Effect, ME)”模型是一个广泛使用的数学模型,被用于量化数据集解决某个问题的潜力。在利用宏基因组辅助预测蛋白质结构这一问题下,“边际效应”ME (Bi, Pj) 被定义为给定生态位 Bi 下的宏基因组数据在补充特定蛋白家族 Pj 的同源序列过程中的潜力。越高的边际效应通常表明更高的同源序列。以蛋白家族PF12652 为例,通过边际效应模型估计,发酵罐生态位微生物群落存在该家族多达 6,218 条同源序列,但土壤生态位微生物群落只能找到24条同源序列。实际上,发酵罐和土壤生态位微生物群落中的同源序列补充结果也证实了这种边际效应结果(图 4A)。因此,对于 PF12652,使用发酵罐生态位下的宏基因组可能比土壤生态位下的宏基因组具有更高的补充同源序列的潜力。
接着,我们利用边际效应模型探究四个生态位(肠道、土壤、湖泊、发酵罐)的微生物组数据补充8,700 个未知结构的 Pfam 家族的同源序列的潜力。结果显示,土壤生态位下的宏基因组对大部分的Pfam蛋白家族的边际效应值高于其他生态位的宏基因组。然而,并不是在所有的蛋白质家族中,土壤微生物群落的边际效应值都最高。其他生态位比如发酵罐生态位,对某些蛋白质也有高于其他生态位的边际效应值(图 4)。该结果也再次从进化角度提示,不同生态位中的宏基因组序列具有不同的进化规律。
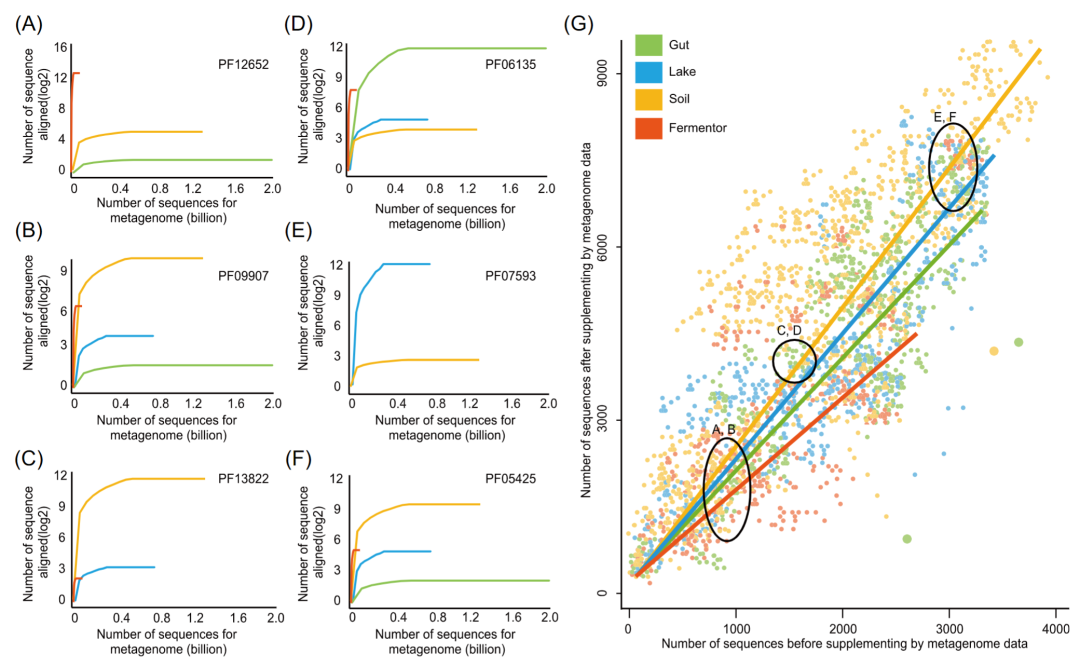
图 4. 边际效应模型评估微生物组辅助预测蛋白家族结构的潜力
该研究评估了四种生态位(肠道、湖泊、土壤、发酵罐)的微生物组数据对所有 8,700 个未知结构的 Pfam 家族(发布版本32.0)的边际效应。不同的颜色表示特定生态位。边际效应模型结果表明,不同生态位对特定 Pfam 蛋白家族的同源序列补充能力可能截然不同。
影响使用宏基因组数据辅助预测蛋白质结构成功的其他因素
从进化的角度,使用宏基因组数据辅助蛋白质结构预测的方法是一种“尽可能发现更多的同源序列”的策略。因此,足够的同源信息,即高质量的多序列比对(MSA)将是蛋白质结构预测成功的关键。作为生成 MSA 的重要参数,选择最佳的序列距离信息(e-value)将在挖掘同源序列时减少 MSA 中包含的噪声序列,从而影响 MSA 的质量,进而影响蛋白质 3D 结构建模的成功。为了预测在这个过程中的最佳e-value,Yang等人设计了一个e-value的预测模型,用于构建 MSA 时选择最佳e-value,减少噪音。
从生态的角度,之前的研究已证明,每个生态位都富含一组特定的物种。而这一特性影响了蛋白质结构预测的结果:为了适应不同生态位下独特的环境压力,该生态位下富含的微生物必须进化出独特的功能基因,以在该特定生态位中获得优于其他物种的生存优势,因此某些功能基因(或蛋白质家族)极有可能在特定生态位中富集。
非靶向和靶向方法来利用宏基因组辅助预测蛋白质结构
如今已经有许多利用宏基因组辅助预测蛋白质3D 结构流程(表 1)。随着宏基因组序列数量的迅速增加,如何有效的利用宏基因组预测蛋白结构变得越来越重要。而回答该问题的关键指标是上述的宏基因组利用效率和边际效应模型:对于特定的蛋白质,宏基因组数据集具有越高的宏基因组利用效率和边际效应,就越能够高效地预测蛋白质3D 结构。为提高微生物利用效率,针对是否充分了解宏基因组和预测出的蛋白结构之间的关联性,预测方法被分为非靶向方法和靶向方法。
非靶向方法(图 5 A)没有利用宏基因组数据与预测蛋白质之间的关联。于是在搜索同源序列时,只能盲目的搜索,对微生物数据的搜索空间没有限制。因此,非靶向方法对生态位微生物群落数据集的利用效率低下。同时,非靶向方法缺乏过程可控性和结果可解释性。
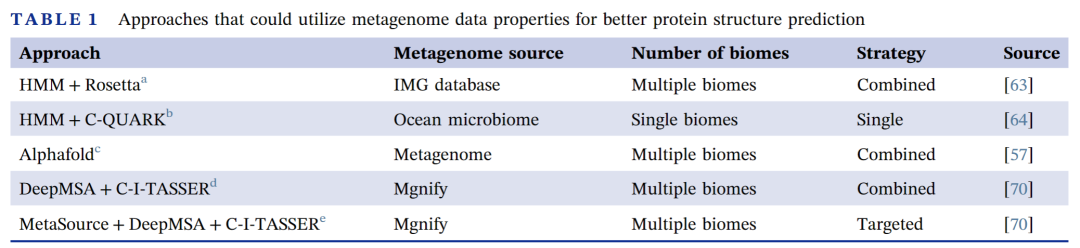
表 1.利用宏基因组数据辅助预测蛋白质结构的方法汇总
在汇总的方法中,“single”表示使用单一生态位的宏基因组搜索蛋白质同源序列。“Combined”表示使用多个生态位的宏基因组数据搜索蛋白质同源序列。“Targeted”表示针对不同蛋白质,选择不同生态位的定制方法。
靶向方法利用宏基因组分布特性来更好地预测蛋白质结构
与非靶向方法相比,靶向方法(图 5 B)利用宏基因组序列和蛋白质家族之间的相关性,有针对性的而不是盲目搜索,从预测出的存在最多同源序列的生态位来补充其同源序列。通过限制蛋白质序列搜索空间的方法,减少宏基因组搜索空间和时间,提高宏基因组利用效率。
靶向方法的思路是:利用宏基因组序列和蛋白质家族之间的相关性,为给定的蛋白质家族预测一个生态位或一组生态位,从这些选定的生态位中能搜索出比其他生态位更多的同源序列。Yang等人构建的机器学习模型(命名为MetaSource)可以作为靶向方法的代表。MetaSource利用不同的生态位富含不同蛋白质同源序列的事实,可以为指定蛋白质预测出为其提供最多同源序列的生态位。经过验证,使用MetaSource预测的生态位比来自不同生态位组合数据预测的蛋白质模型更高效和准确。
作为一种靶向方法,MetaSource 可用于减少在蛋白质结构预测中补充同源序列的步骤所花费的时间,能直接影响结构预测的效率。评估结果显示,利用MetaSource指定的宏基因组预测蛋白结构时,利用效率为每十亿宏基因组序列有7,810 条同源序列,这比使用 IMG 数据库的微生物利用效率(每十亿宏基因组序列 160 个同源序列)高出 50 倍(图 3 B)。
本工作使用PF07682 和 PF05005这两个具有已知结构的Pfam蛋白家族来验证靶向方法的高效性和准确性(图 5 C)。经过验证,尽管来自组合生态位(使用不同来源的生态位)搜索到的同源序列比单个生态位多,但来自组合生态位的结构模型不如某些单个生态位(土壤或湖泊)的结构准确,这很可能是由于MSA中的噪声序列导致的(图 5 C)。作为靶向方法,MetaSource 预测出的生态位(PF07682:土壤生态位,PF05005:湖泊生态位)可以减少噪音序列,辅助构建出和天然结构最相似的蛋白质 3D 结构,并且使用的宏基因组序列比非靶向方法少得多。造成这种情况的原因可以用蛋白家族的物种组成解释:PF07682 和 PF05005 主要由来自Proteobacteria和Cyanobacteria的蛋白质组成,它们分别在土壤和湖泊生物生态位中占主导地位。这一结果支持了靶向方法的优势:高精确度、高效率和可解释性。
总之,从生态和进化的角度,宏基因组利用效率和边际效应是利用宏基因组数据有效预测蛋白质结构的关键指标。分析结果表明,边际效应分析得出不同生态位中的微生态位对不同蛋白家族的补充潜力是截然不同的,所以宏基因组利用效率高度依赖数据和方法的选择。在数据方面,它严重依赖于选择的生态位中的蛋白质组成;在方法方面,非靶向方法和靶向方法将导致宏基因组利用效率的截然不同。此外,在许多情况下,靶向方法会产生更精确的蛋白质结构,因为所涉及的噪音序列更少,正如两个示例的Pfam蛋白家族的结果所证明的那样。
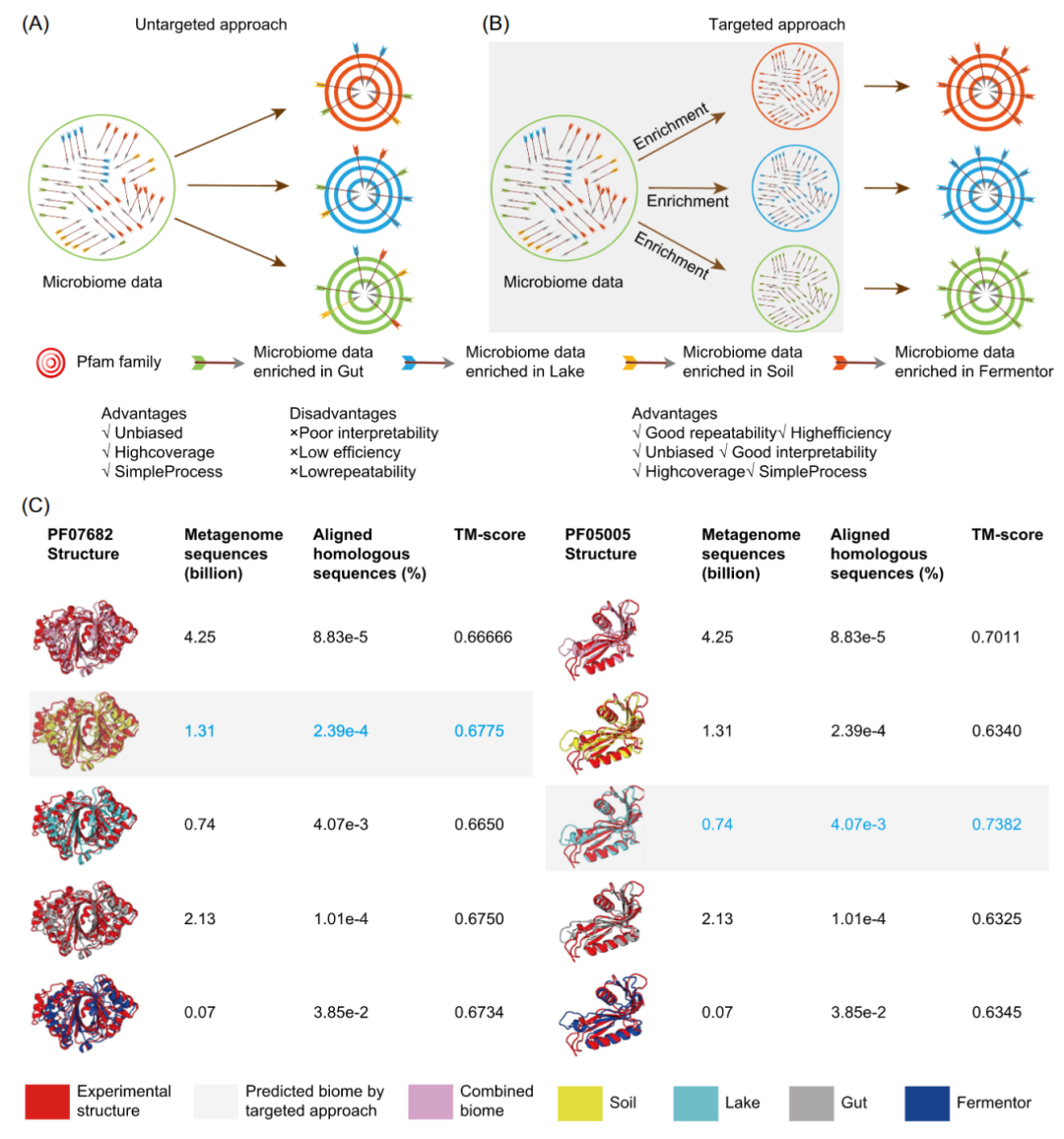
图 5. 靶向方法本质上是一种检测生态位中富集蛋白的方法
在利用宏基因组辅助预测蛋白质 3D 结构的过程中,(A)非靶向方法盲目地搜索宏基因组数据,不限制搜索空间。(B) 靶向方法只用搜索特定生态位的宏基因组,限制搜索空间。靶向方法根据宏基因组数据与蛋白结构之间的关联性,预测出能够补充最多同源序列的生态位下的宏基因组。 (C) 为了比较靶向方法和非靶向方法,使用来自不同生态位的 MSA为Pfam 蛋白家族PF07682 和 PF05005 进行模型构建。对于每个生态位,计算搜索到的同源序列占所有宏基因组序列数量的比例作为微生物组利用效率。通过将预测出的3D结构与已知结构进行比较,并利用TM-score量化两者的相似性。MetaSource 是在先前的研究中开发的靶向方法。标有红框的模型是 MetaSource 预测的含有最多同源序列的生态位。被标为蓝色字体的模型是具有最高 TM-score的模型。
探究蛋白质结构预测所需的宏基因组数据的下限
不依赖于模板的蛋白结构预测方法需要大量同源序列,所以补充同源序列对构建蛋白质的可靠结构将是至关重要的。尽管蛋白质结构预测所需的宏基因组序列的确切下限难以被量化。但这一下限可以基于之前计算过的两个关键指标来估计:宏基因组利用效率和边际效应。在估计下限之前,我们做了一些前置参数的计算:
(1) 在当前 Pfam 数据库中 (http://pfam.xfam.org/),通过统计蛋白质的数量 N(Pj)、蛋白质家族的同源序列Homo(Pj),可以推导出蛋白质的平均同源序列数量:AveHomo(Pj);
(2) 对于当前的宏基因组数据(通过整合 IMG 数据库、Mgnify 数据库和 NCBI SRA 数据库的数据),可以确定生态位的数量 N(Bi);
(3) 基于之前研究中的方法和统计结果,用来自特定生态位 Bi 的宏基因组数据补充特定蛋白质 Pj的同源序列。统计出宏基因组利用效率 UE(Bi, Pj)。结合蛋白质的数量 N(Pj),统计出平均宏基因组利用效率 Ave(UE)(公式1)。
(4) 基于之前研究中的统计结果,可以统计来自特定生态位 Bi 的宏基因组数据在补充特定蛋白质 Pj 的同源序列时的边际效应 ME (Bi, Pj)。基于这些假设,当使用非靶向方法时,可以粗略的估计需要的宏基因组序列总数为(公式2).
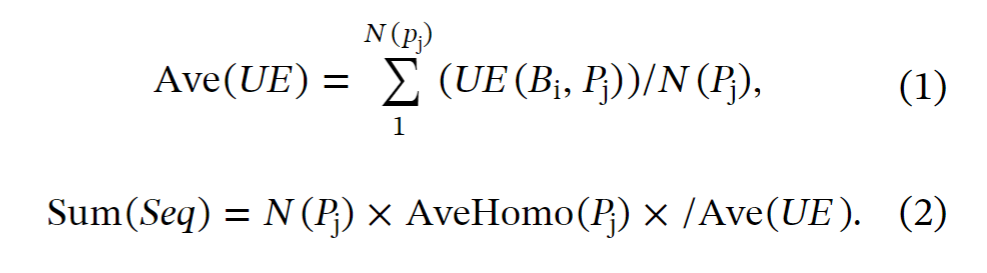
而根据Pfam数据库中的蛋白质同源序列信息的数据统计,蛋白质平均同源序列数量为AveHomo(Pj) ~ 3713, 蛋白质的数量 N(Pj)基于 Pfam 发布版本34.0中的数据,为 19,179。根据本工作的实验结果,平均宏基因组利用效率Ave(UE)约为100(每十亿条宏基因组序列)。因此,基于最保守的估计,需要的宏基因组序列总数Sum(Seq)约为7.12E12。
当使用靶向方法时,同源序列数量的下限可以大大减少。首先,靶向方法的平均利用效率的计算是基于四个代表性生态位(肠道、土壤、湖泊、发酵罐)的微生物组数据。平均宏基因组利用效率(每十亿宏基因组序列):肠道:10,土壤:248,湖:142,发酵罐:320。四个生态位的平均利用效率为每十亿个宏基因组序列180个,相当于(10(肠道)+248(土壤)+142(湖)+320(发酵罐))/4(生态位数量)(公式1)。根据平均宏基因组效率,预测Pfam数据库中的所有蛋白质,需要的宏基因组序列的数量为:
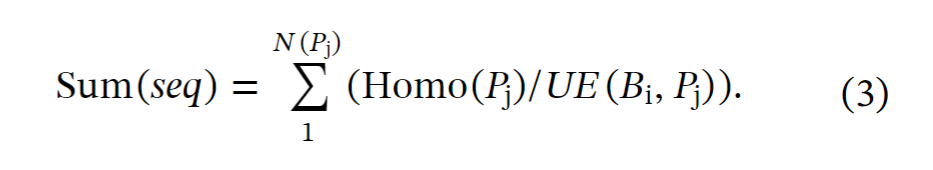
我们可以估计宏基因组的数据下限为 4.32E12,远小于非靶向方法的下限。
更进一步,我们根据不同的Pfam发布版本,探究了蛋白质同源序列的增加和所需宏基因组序列的下限之间的相关性(图 6)。结果显示,随着 Pfam 数据库中序列数量的增加(图 6A),所需宏基因组序列与目前已有微生物组数据之间的差距正在扩大(图 6B)。鉴于当前的 Pfam 数据库有 19,179个蛋白质家族,估计有 7.12E12 条宏基因组序列才可以预测所有蛋白质结构,但当前的宏基因组数据库只有大约 1.48E12 条宏基因组序列(统计数据来自三个常用宏基因组数据库:IMG 数据库、Mgnify 数据库和 SRA 数据库)。而由于拥有更高的宏基因组利用效率(靶向方法:每十亿宏基因组序列搜集到185条同源序列;非靶向方法:每十亿宏基因组序列 100 条同源序列),靶向方法比非靶向方法所需的宏基因组下限要低(根据公式 3, 估计下限为 4.32E12)。说明靶向方法可以通过提高宏基因组利用效率部分解决这一差距。
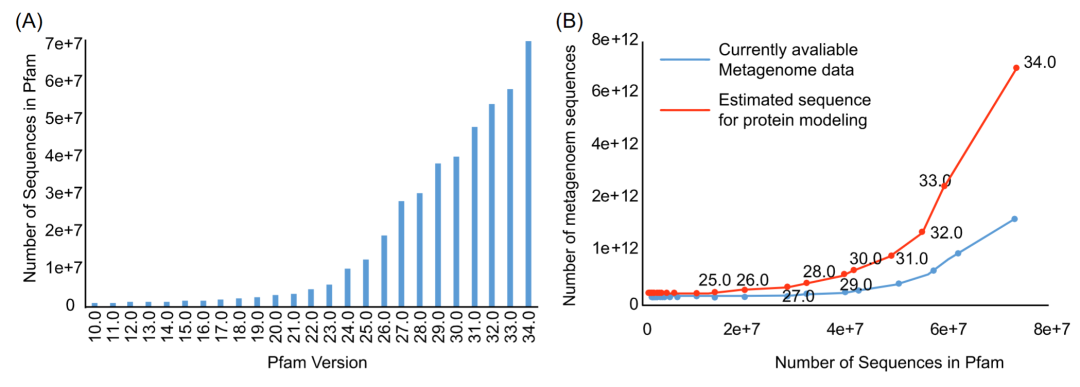
图 6. Pfam数据库中蛋白质数量增加与宏基因组序列数量增加之间的关系
A. 不同Pfam 版本中的蛋白序列数。B. 预测Pfam中所有蛋白序列所需的宏基因组序列数与目前测序得到的宏基因组序列数的关系。每个节点代表一个 Pfam 发布版本。红色的曲线模拟的是计算后预测Pfam中所有蛋白需要的宏基因组序列数目。蓝色的曲线模拟的是同一时间节点下,实际上搜集到的微生物组序列数
结论与讨论
利用宏基因组序列辅助预测蛋白质 3D 结构并解码蛋白质结构和功能是一种非常有前景的方法。而本工作揭示了宏基因组序列辅助预测蛋白结构背后的数据依赖和方法依赖模式:来自不同生态位的宏基因组序列对特定蛋白质家族的贡献截然不同,而利用这种差异分布的靶向方法比非靶向方法能更有效地补充同源序列并获得更精确的蛋白结构。
蛋白质家族同源序列的有效补充问题本质上是对蛋白质生态和进化模式的探究:为了适应生态位的特定环境压力,微生物中适应环境压力的功能基因会进化,从而使其宿主物种能够获得生存优势。因此某些功能基因(或蛋白质家族)极有可能在特定生态位中富集。
在生态模式方面,在蛋白质家族中寻找同源序列的本质实际上是一个富集问题:我们可以从哪个生态位中最有效地挖掘同源序列。我们的评估结果已经表明,像 MetaSource 这样的靶向方法可以利用微生物生态位与同源序列之间的联系,使我们能够从特定生态位微生物群落中推断出富集的功能基因。这也在一定程度上解释预测出的蛋白质在来源生态位中的重要作用,增加整个过程的可解释性。
在进化模式方面,靶向方法可以预测蛋白质的来源生态位,以找到足够的进化信息(即同源序列)来预测其可靠结构。与仅提供宏基因组中现有进化信息的非靶向方法不同,靶向方法还可以指导寻找自然界中尚未测序的同源序列:即使在目前的宏基因组中不能为蛋白质提供足够的同源序列,我们可以通过测序靶向方法预测的具有最多同源序列的生态位下的宏基因组样本来补充同源序列。
此外,我们估计了预测Pfam 数据库中所有蛋白质的 3D 结构需要多少宏基因组序列,而我们发现当前的宏基因组数据无法满足这一需求。一方面,收集更多的宏基因组序列是一种解决方法;而另一方面,我们又需要减小搜索空间来平衡预测能力和分析效率。针对这两个方面的矛盾,靶向方法将是理想的方案,因为它可以通过利用生态位内部的生态和进化信息,提高宏基因组使用效率,指导后续的同源序列补充来缩小这一的差距。
总的来说,靶向方法(以 MetaSource 方法为例)利用不同的生态位富集不同的蛋白质这一事实,极大地提高了宏基因组数据的利用效率,在使用宏基因组序列预测蛋白质结构方面具有巨大潜力。本工作认为靶向方法是从宏基因组序列预测蛋白质结构的双赢解决方案:一方面,它不仅可以大幅度减少需要搜索的序列的数量,也可以改善许多蛋白质家族的预测精度。另一方面,靶向方法提供了有关蛋白质的生态和进化规律的丰富知识。
引文格式:Pengshuo Yang, Kang Ning. 2022. How much metagenome data is needed for protein structure prediction: The advantages of targeted approach from the ecological and evolutionary perspectives. iMeta 1: e9. https://doi.org/10.1002/imt2.9
作者简介

杨朋硕
● 华中科技大学创新研究院博士生
● 2017年本科毕业于华中科技大学,之后在生命科学与技术学院宁康教授的指导下攻读博士学位。以第一作者身份发表SCI论文4篇 (Genome Biology, 2019; PNAS, 2021等),并参与五项国家自然基金委项目(其中一项为第二完成人),七项发明专利。研究方向为生物大数据挖掘,以及微生物组学研究中的方法开发。重点关注环境和健康领域的生物大数据处理。

宁康(通讯作者)
● 华中科技大学生命科学与技术学院教授,博士生导师,生物信息与系统生物学系系主任
● 2008年博士毕业于新加坡国立大学计算机学院生物信息专业。在生物信息学和微生物组学领域从事科研工作10余年,作为通讯作者在PNAS、Gut、Genome Biology、iMeta、Gut Microbes、Briefings in Bioinformatics、Bioinformatics等生物学、医学和生物信息学顶级学术期刊等高水平学术期刊发表学术论文60余篇,文章总引用超过3000次,H指数30(Google Scholar)。目前主持国家自然科学基金项目、科技部重大研究计划课题等。担任中国生物信息学学会(筹)-基因组信息学分会副主任,中国生物工程学会-计算生物学与生物信息学专业委员会委员,中国计算机协会-生物信息学专业委员会委员等。担任iMeta、Genomics Proteomics Bioinformatics、Microbiology Spectrum、Scientific Reports等国际期刊编委。
详细情况请参见:http://www.microbioinformatics.org/
更多推荐
(▼ 点击跳转)
iMeta | 南科大宋毅组综述逆境胁迫下植物向微生物组求救的遗传基础(附招聘)
iMeta:青岛大学苏晓泉组开发跨平台可交互的微生物组分析套件PMS

iMeta:哈佛刘洋彧等基于物种组合预测菌群结构的深度学习方法
iMeta:吴青龙/王明福/刘金鑫等-从肠道菌群看待人类对高原饮食的适应性
iMeta:西农韦革宏团队焦硕等-土壤真菌驱动细菌群落的构建
期刊简介

“iMeta” 是由威立、肠菌分会和本领域数百位华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表原创研究、方法和综述以促进宏基因组学、微生物组和生物信息学发展。目标是发表前10%(IF > 15)的高影响力论文。期刊特色包括视频投稿、可重复分析、图片打磨、青年编委、前3年免出版费、50万用户的社交媒体宣传等。2022年2月正式创刊发行!
联系我们
iMeta主页:http://www.imeta.science
出版社:https://onlinelibrary.wiley.com/journal/2770596x
投稿:https://mc.manuscriptcentral.com/imeta
邮箱:office@imeta.science

微信公众号
iMeta
责任编辑
微微
往期精品(点击图片直达文字对应教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|




























后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集




















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









