






点击蓝字 关注我们
海洋RNA病毒圈的二十年研究

https://doi.org/10.1002/imt2.59
REVIEW ARTICLE
● 2022年10月17日,中国科学院上海巴斯德研究所崔杰团队在iMeta在线发表了题为“Over two decades of research on the marine RNA virosphere”的文章。
● 该文章回顾了海洋RNA病毒信息的分类学发展,总结了RNA病毒组学的研究方法及其主要进展,为今后开发探索海洋RNA病毒圈的进化起源和生态意义提供方向。
● 第一作者:廖蒙恩、谢蕴怡
● 通讯作者:崔杰 (jcui@ips.ac.cn)
● 合作作者:施莽
● 主要单位:中国科学院上海巴斯德研究所
亮 点
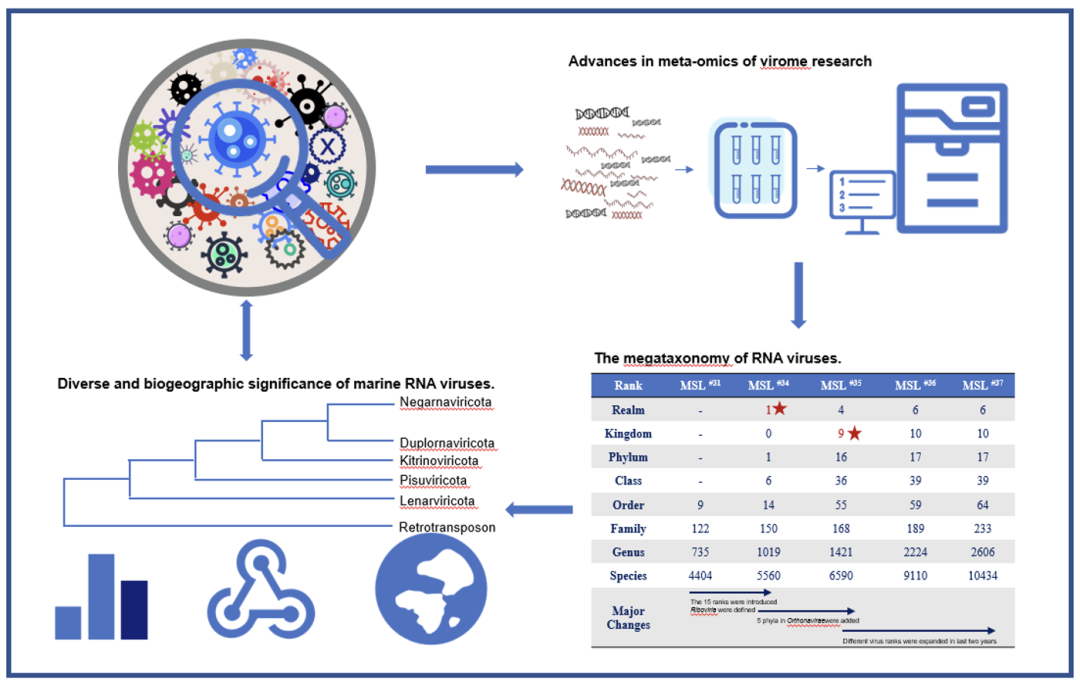
● 宏组学技术的发展提供了大量海洋RNA病毒信息
● 深海病毒的发现极大地促进了RNA病毒的分类学发展
● 了解海洋RNA病毒的多样性和生物地理学有助于揭示其进化轨迹、分级分类和它们在生态系统的重要性
摘 要
RNA病毒(域:Riboviria)包括RNA噬菌体和感染真核生物的RNA病毒,是海洋生态系统的重要组成部分。在过去的二十年中,随着第二代测序(NGS)技术的快速发展和宏组学方法在病毒学研究中的应用(即病毒组学,对一个特定的环境中所有病毒的研究),科学家们发现了大量的海洋RNA病毒,以此对RNA 病毒的分类学和遗传多样性也有了更深的认识,并发现RNA病毒对复杂的生态系统有着多种重要影响。此外,通过比较不同深度海洋生态环境中的病毒组,科学家们揭示了海洋RNA病毒的生物地理模式。本篇综述中,我们总结了全球海洋RNA病毒圈的特征,并重点概述了RNA病毒的进化分类研究历史。除此之外,我们还总结了RNA病毒组学的研究方法及其主要进展。本综述不仅是为了深入了解海洋RNA病毒,还为今后开发更好的研究方法和探索海洋RNA病毒圈的进化起源和生态意义提供方向。
关键词:生态学,海洋RNA病毒,宏基因组学,宏转录组学,分类学,病毒进化
视频解读
Bilibili:https://www.bilibili.com/video/BV15G4y1H7GT/
Youtube:https://youtu.be/b0qrKT1pEHY
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
引 言
最早记录的海洋病毒是于1966年在一种海蟹(Liocarcinus depurator)中发现的。从那以后,人们在探索海洋中的病毒病原体上做了大量工作。水生环境中分布着大量病毒,每毫升海水中存在2.5 × 108个病毒颗粒,它们可以通过裂解细胞和参与地球生物化学循环。自然环境中的DNA病毒的研究很丰富,但我们对海洋生态系统中RNA病毒的特征(丰度、流行率、分布、生态学以及进化模式和轨迹)知之甚少。最近,探索全球RNA病毒圈的研究揭示了它们在自然环境中的功能和重要性。
海洋占地球表面70%以上的面积,具有独有的特征(如缺氧、光照有限、静水压力高和无机化合物浓度高),为水生生物提供了一个复杂且多变的环境。海洋根据深度不同可以细分为五层(从海面到海底依次为上层、中层、深海层、深渊层和深海层区),由于阳光照射不均,它们的环境有很大差异。科学家们已经揭示了海洋RNA病毒圈在不同生物地球化学层之间和内部的相似性和异质性,并试图预测这些关系随着环境因素而变化的方式。自20世纪90年代初,人们还发现病毒除了作为海洋生物的病原体外,还能通过影响宿主群落和生物地球化学循环来影响海洋生态系统。例如,病毒可以通过生物泵作用将有机物从上层运送到深层,并且病毒通过裂解宿主,在全球碳循环中发挥作用。最近的一项研究发现海洋碳循环与RNA病毒之间的潜在联系,它们可能作为生物泵的一个重要组成部分感染海洋宿主,并参与编码辅助代谢基因(AMGs,如负责营养物质运输和光合作用的基因)。除此之外,通过一些特定的海洋RNA病毒的丰度还可能预测海洋碳通量。
随着高通量测序技术的发展,现在科学家们可以通过一系列不依赖培养病毒的方法来探索海洋病毒。2015年,一次全球科学考察报告了5476个海洋病毒种群,其中大部分是噬菌体,这一数字在2016年扩大到15222个,2019年扩大到195728个。公共数据库中的RNA病毒信息量剧增,随之而来的挑战是确定其分类。依靠RNA依赖的RNA聚合酶(RdRp)的比较和系统发育分析可以在一定程度上解决这个问题。由于RdRp起源古老和突变率高,不同RdRp序列分歧度可能较大,使用RdRp蛋白质结构比较和隐马尔可夫模型(HMM)等有助于解决由此引起的问题。
由于取样和培养困难,我们仍未明晰海洋RNA病毒的全貌,对海洋RNA病毒在特定生态环境中的生态学意义仍知之甚少。因此,我们对海洋RNA病毒的研究不能仅局限在其临床和经济影响,而需要全球范围的广泛研究。本综述总结了我们目前(特别是自第二代测序(NGS)技术出现以后)的对RdRp编码的海洋RNA病毒(界:Orthornavirae)的认识,强调了它们的基本特征和适应宏组学时代的新型分类系统(图1)。最后,我们讨论了研究海洋RNA病毒圈的理论和方法,以及从不同角度看未来的挑战和方向。
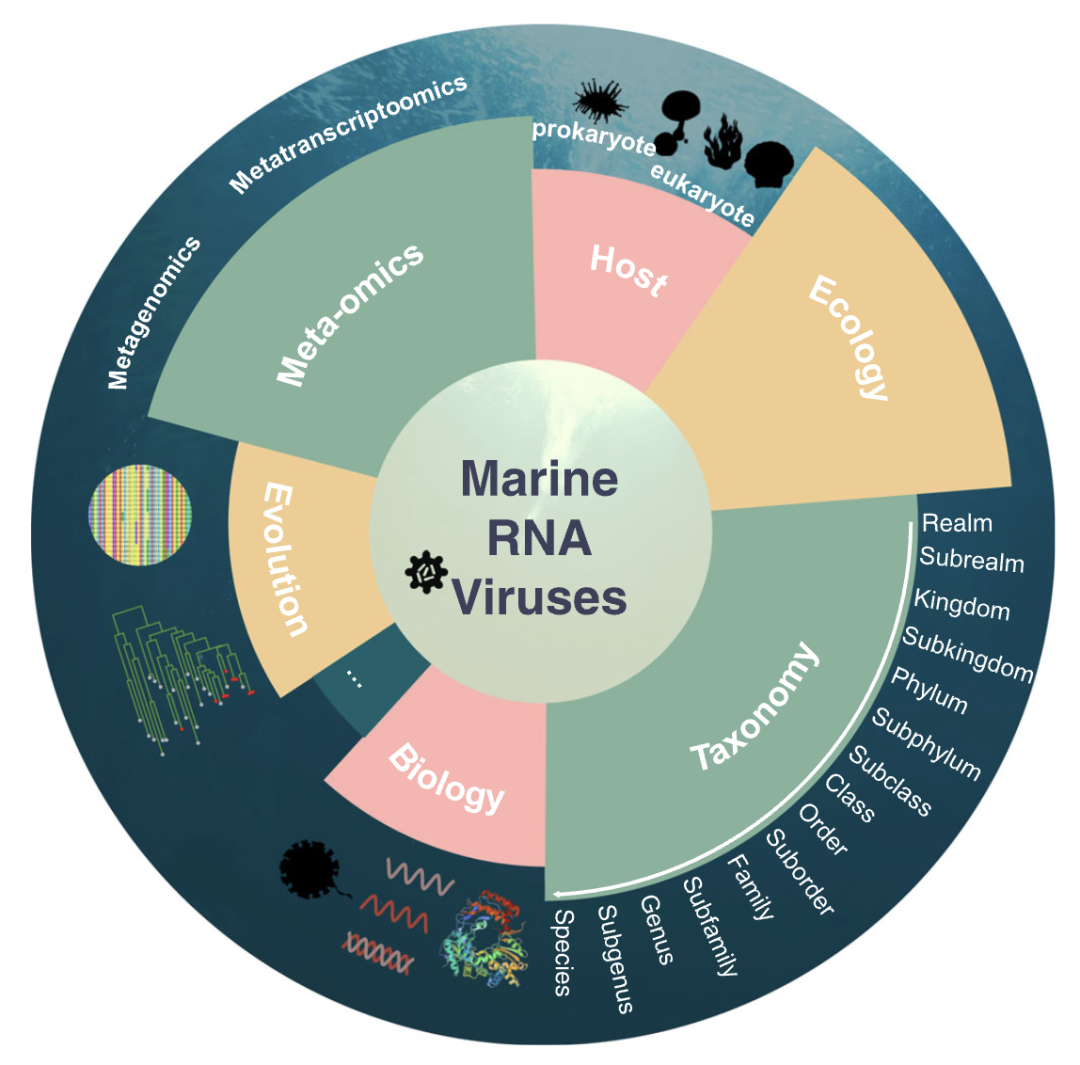
图 1. 海洋RNA病毒的不同方面
在描述海洋RNA病毒的生物学和流行病学特征以及宿主范围方面,科学家们已经做出了许多尝试。如今,由于宏组学(包括宏基因组学和宏转录组学)的出现和发展,大量的海洋RNA病毒被发现,促进了RNA病毒圈的分级分类和深度进化研究。此外,随着全球采样和比较研究的发展,海洋RNA病毒的生态学意义也逐渐清晰。该分类法来自于国际病毒分类委员会(ICTV)网站(https://ictv.global/taxonomy)
海洋RNA病毒的特点
1. 生物学特征
海洋RNA病毒间具有复杂的基因组特征和形态学特征。它们的基因组承载着生物体的遗传信息,病毒颗粒的形态结构对于研究病毒的感染机制具有重要意义。
RNA病毒以RNA作为遗传物质。它们的基因组有多种复杂形式,如分段、非分段和环形基因组。通常来说,RNA病毒可分为单链RNA(ssRNA)病毒和双链RNA(dsRNA),它们之间的主要区别是它们的基因表达模式不同。RdRp作为所有RNA病毒中唯一保守的序列,是几种聚合酶的祖先和肽-RNA世界中最早的基因之一,广泛用于研究RNA病毒的起源。RNA病毒基因组的大小通常在4kb到12 kb之间,已知最大的RNA病毒基因组是冠状病毒的基因组(41 kb)。限制RNA病毒基因组大小的主要因素是其复制保真度低,RdRp的每一个核苷酸引入大约10-4个错误复制,这一特性使RNA病毒能够在很短的生成时间内产生具有不同病毒变体,导致RNA病毒的基因组中的基因重叠,从而使基因组压缩。此外RNA病毒复制过程中缺乏校对、修复和复制后纠错机制。因此,RNA病毒基因组不稳定,容易发生突变,常常表现出频繁的病毒-宿主重组、水平基因转移、基因增减和重排。这种不稳定性加大了RNA病毒研究的难度。
不同科的RNA病毒在形态上有差异,具体可体现在衣壳蛋白和尾部,可以通过定量透射电子显微镜观察到病毒头体直径和尾部长度的差异进行鉴定。病毒衣壳蛋白的自发组装过程是由包膜蛋白上的正电荷和基因组上的负电荷之间的静电相互作用驱动,这种组装使衣壳蛋白二级/三级/四级结构高度有序,从而构成了病毒衣壳的不同形态,如球形/二十面体和圆柱形/螺旋形。科学家们通过人工设计的病毒衣壳蛋白对RNA进行封装,可以作为保护和传递RNA信息的一种新手段。之前的研究表明,无尾病毒在地表水中占主导地位(平均占所有病毒的79%),RNA病毒可能占观察到的无尾病毒的16%-100%。但由于从海洋环境中分离和培养RNA病毒困难,对海洋RNA病毒形态的研究不多,因此对RNA病毒的形态的更多认识仍需科学家们更多的探索。
2. 海洋RNA病毒的宿主
病毒依赖于宿主存在,理论上它们可以侵染任何细胞形态的生物。病毒感染宿主后,可以直接裂解宿主细胞进行繁殖,也可以形成将病毒基因整合进宿主基因中的RNA噬菌体(仅在原核生物中),随宿主基因复制遗传。目前鉴定组学分析出来的RNA病毒的宿主具有一定难度,已有方法通常有如下几种:通过实验培养;通过对数据集比对进行合理预测,如宿主和病毒是否包含一致的成簇规则间隔短回文重复(CRISPR)序列;病毒基因组中存在的编码tRNAs和AMGs的序列来源;其他病毒基因组片段和潜在宿主基因片段之间的相似性来确定宿主;样本中病毒和潜在宿主的丰度和表达水平来预测宿主;病毒和潜在宿主之间的相似四核苷酸频率。RNA病毒与宿主间存在多样的相互作用,宿主中的抗病毒限制因子可结合RNA病毒,从而抑制病毒的复制和表达。而在受感染的宿主中,RNA病毒可以诱导产生降解限制因子的蛋白以抑制宿主基因表达。除此之外,RNA病毒还可以诱导宿主表达前病毒复制RNA,如lncRNA-ACOD1等。RNA病毒与宿主的相互作用在理解病毒进化和生态变化方面起着至关重要的作用。原生动物、动物和植物等宿主中共享的RNA病毒基因序列的存在解释了不同宿主之间的基因水平病毒转移是RNA病毒进化的重要方式。RNA病毒与其宿主的共分离为跨物种传播提供了基础,并为RNA病毒的宏观进化提供了解释。病毒与宿主的相互作用还可能会影响生态系统、疾病传播和其他与人类生活相关的领域。例如,病毒与宿主之间的相互作用可以控制原核生物的死亡,释放惰性有机物,补偿深海生态系统中被抑制的宿主代谢途径;在深海热液喷口,RNA病毒可以裂解宿主细胞,以控制宿主种群的规模,并通过杀死特定的微生物来影响宿主群落结构。
3. 海洋环境中RNA病毒的丰度
病毒是生态系统的重要组成部分,广泛存在于土壤、人类和海洋等环境中。RNA病毒感染影响人类健康,海洋RNA病毒感染也影响着海洋健康,海洋中丰富的RNA病毒重塑了我们对病毒早期进化的理解。破译海洋中病毒的作用、行为和功能可能是“本世纪最大的谜团之一”。温度、洋流、宿主分布和生物地理变化都会影响宿主丰度和病毒丰度。在海洋环境中,从低生产力系统到高生产力系统,RNA病毒从每升108个病毒颗粒到超过1011个病毒颗粒不等。RNA病毒的分布还受到海洋纬度的影响,例如,Taraviricota在温带和热带地区的丰度很高,而负ssRNA门“Arctiviricota”在大西洋北极水域的丰度最高。海洋生物污损会对生物多样性和海洋生态系统造成破坏,RNA病毒可通过这些生物污染传播到全球海洋。大量RNA病毒的传播会带来疾病,影响人类的海洋活动,并危及人类的可持续发展。海洋中有着丰富的RNA病毒,由于对许多RNA病毒的宿主采样不足,导致环境RNA病毒丰度测量的局限性,解决这个问题将需要更多的病毒和样本数据。
4. 病毒在海洋环境中的适应性
极端的海洋环境,如深海、极地、高温地区、热液喷口以及高压或盐度高的地区,都接近生命生存环境的极限。病毒群落可以通过扭曲特定氨基酸残基的编码频率来适应极端环境。RNA病毒本身具有的高突变性特点可以帮助它们适应各种环境,极端环境不会增加病毒的突变率,但来自当地环境的选择压力可以增加某些类型病毒的丰度。RNA病毒的高度适应能力有助于产生由具有广泛表型和遗传异质性的突变谱组成的群体。当环境适宜生存时,在极端环境下占主导地位的突变变体可以保留为少数变体,当病毒再次暴露在相同环境时,这些变体可以快速选择繁殖。故而RNA病毒能适应不断变化的选择压力,在多变的海洋环境中生存。极端海洋环境的独特生态压力还可能使病毒及其宿主微生物群落合成具有不同生物活性的新化合物,这些化合物在生物地球化学循环中发挥着重要作用。
深海海洋病毒的发现有助于RNA病毒的分类研究
为了了解感染人类、感染具有经济或社会意义的动物和植物的致病病毒,人们做出了大量研究。病毒在生物圈广泛分布,全球公共数据库中已有极为丰富的病毒数据,远远超出了病毒学家在实验室中所发现的病毒量。因此,在过去20年里,病毒分类学不断发生变化,RNA病毒分类学也有了巨大变革(表1)。
表1,RNA病毒的分类学历史和更新

来源于International Committee on Taxonomy of Viruses (ICTV) Master Species List 2016 v1.3, https://talk.ictvonline.org/files/master-species-lists/m/msl/6776
1. 早期分类——病毒的五个分类
病毒通过不同的策略复制和表达它们的基因组,1971年巴尔的摩基于这些策略将所有已知的病毒分为六组,后来又引入第七组。巴尔的摩分类系统基于病毒核酸的形式、极性和表达,将RNA病毒分为dsRNA病毒、正单链RNA、负义单链RNA和RNA逆转录病毒,这一分类系统至今仍然被广泛使用。然而,最近的研究表明,同一巴尔的摩分类的病毒并不一定起源于同一个共同祖先,这说明该分类系统不一定反映了不同群体内部和不同群体之间的实际进化关系。
最早的病毒研究主要通过实验室分离和培养病毒。1970-1990年间,国际病毒分类委员会(ICTV)对一个新病毒物种的标准主要取决于病毒的生物学特性,如其在体外的特性、病毒颗粒结构和抗原关系,同时,也要考虑许多宿主因素(如毒力、致病性、宿主范围和流行病学)。在此基础上,ICTV将病毒分为两个不同的级别,随后将病毒分类系统扩大到包括种、属、亚科、科、目五个等级的结构。这个等级结构一直沿用到2017年,当时一共包括9个目、122个科(37个亚科)、735个属和4404个种 (ICTV Master Species List 2016 v1.3, https://talk.ictvonline.org/files/master-species-lists/m/msl/6776)。
2. 核糖病毒域的5分支结构和15个层次的分级
随着Sanger测序技术的进步和病毒遗传学的发展,病毒基因组测序技术逐渐成熟。因此,通过序列差异和系统发育的病毒序列推断,结合实验和流行病学因素可以作为病毒分类的标准。例如,Heterosigma akashiwo RNA病毒(HaRNAV)是在不列颠哥伦比亚省沿海地区分离出的第一个海洋RNA病毒,能感染可以形成红潮的有毒光合藻类H. akashiwo。根据HaRNAV的宿主、形态、病毒颗粒大小、基因组大小、特有结构域以及与近缘病毒的系统发育关系,将其定义为Picornavirales病毒目中新命名的Marnaviridae病毒科的第一个成员。
鉴于病毒,特别是对于处于不同级别的病毒和不同巴尔的摩类的病毒,缺乏可用于构建包含所有病毒的统一系统发育树的共同标志基因,病毒的分类学在一定程度上与细胞生物的分类学不同。对于RNA病毒来说,同源的RdRp的存在可以推断出序列的距离和病毒科之间的进化关系,这为新的分类提供方法。
在宏基因组学时代,环境或生物体中与已知病毒相似度有限或无法分类的潜在病毒超过了ICTV公布的病毒数量。在一项海洋和内陆无脊椎动物的宏转录组研究中确定了1445种RNA病毒,其中一些与已知的病毒科有明显的差异。因此,ICTV曾主持专题会议讨论是否以及如何将宏因组学发现的病毒纳入ICTV分类法,并达成了一个共识,即拟议的分类法可以应用于病毒基因组和宏基因组序列数据。另外,从基因组分析中预测的表型和从序列比较中推断的距离可以作为病毒分类学的基本要素,生物学数据不再是必不可少的。2016年,ICTV工作组讨论并批准了15级病毒分级,将全球病毒圈分为8个一级分类和7个二级分类的分类模式,作为结合林奈分类系统和巴尔的摩分类学的新型病毒分类系统。一级分类包括四个已经存在的等级(目、科、属、种)和四个新纳入的等级(域、界、门、纲),而二级分类包括以前使用的亚科等级和六个由一级等级衍生的新等级(亚域、亚门、亚界、亚纲、亚目和亚属)。随着病毒发现的持续进行,这一更新的分类系统既可作为新发现病毒分类的动态框架,也可以涵盖许多仍在原病毒分类系统之外的病毒。
后来建立的病毒域,即Riboviria,包括来自三个巴尔的摩分类的编码RdRp的RNA病毒(https://talk.ictvonline.org/taxonomy/p/taxonomy-history?taxnode_id=202107095)。为了重新研究RNA病毒的进化关系,研究人员开展了比较基因组研究,将RNA病毒RdRp系统发育分类为五个主要分支。在拟议的Orthornavirae界(编码RdRp的RNA病毒,与编码RT的RNA病毒相对)中,这五个分支为Lenarviricota(分支1)、Pisuviricota(分支2)、Kitrinoviricota(分支3)、Duplornaviricota(分支4)和Negarnaviricota(分支5)这五个门。为了解决RdRp序列的高分歧度和人工整理过程中的主观性等问题,一些补充性方法,如使用RdRp核心序列、HMM模型和RdRp三维结构进行补充分析,结果支持基于RdRp的序列分类,但也有一些修改和补充。Riboviria的建立和随后的5个分支的病毒分类划分被认为是应用新提出的病毒分类等级结构的第一步,进一步帮助阐明了RNA病毒之间的关系,有利于新出现的RNA病毒的发现、分类和命名。
3. 全球RNA病毒拟议分类的10个门
为了进一步揭示海洋病毒圈,科学家们进行了更多的大范围研究。在全球海洋RNA病毒多样性的最新研究中,Ahmed A. Zayed及其同事通过优化的迭代搜索方法,从全球收集的771个Tara海洋宏转录组资源和143个来自北冰洋的新宏转录组数据点中恢复了44000多个编码RdRp的组装序列和6686个完整或接近完整的RdRps。除此之外,这项研究表明,六个新建立的宏观分类可能对应五个新的门(命名为 "Arctiviricota"、"Paraxenoviricota"、"Pomiviricota"、"Taraviricota "和 "Wamoviricota"),使正RNA病毒界的已知门类数量翻倍。
此外,根据系统发育、结构和基因组水平的分析,科学家们对已报道的正RNA病毒界的起源和早期关系进行修订,从而更加确认了现有的序列推断的系统发育分类。这些分析表明,Duplornaviricota门(dsRNA病毒)中的病毒具有多起源性,而之前的一项研究表明,这些dsRNA病毒是从(+)ssRNA病毒进化而来的,前者的可能性得到了更多证据的支持。同时,新提出的 "Taraviricota "门,介于逆转录病毒和正RNA病毒界病毒之间,可能是其他正RNA病毒界病毒的早期进化起源。
尽管样本类型偏差仍然存在,但对该领域的研究方法和认知水平的进步,特别是在深层宏基因组学和进化病毒学方面,为全球病毒的发现和分类提供了启示,并揭示了海洋RNA病毒圈的奥秘。
基于宏组学的病毒发现和注释
1. 早期的研究方法
关于海洋RNA病毒的研究报道,最初聚焦于一些感染水产养殖中有重要经济意义的海洋动物的RNA病毒。陆续在海洋中发现了几种感染原生生物的RNA病毒,并发现其宿主范围很广。尽管这些病毒学研究具有指导性意义,但由于主要关注病毒的分子特征,在海洋RNA病毒的生态学(包括其多样性、生物地理学、与宿主的相互作用、进化状况和环境贡献)上仍有缺陷。
随后出现了通过分离病毒的方法来发现环境中的RNA病毒,揭示海洋中RNA病毒群体的多样性。但是针对广泛保守的标志基因的聚合酶链反应(PCR),还是可以在一定程度上应用于发现不同的病毒群体。早在2003年,针对RdRps的引物和反转录-PCR(RT-PCR)技术就已经用于研究海水中picorna-like病毒的存在和多样性。通过序列分析,研究人员确定了四个单系的picorna‐like病毒,可能代表了至少两个新的病毒科,显示了在海洋环境中发现RNA病毒的单基因的潜力。除了在病毒发现方面的贡献,基于RdRp靶向RT-PCR的研究还通过结合采样位置和时间,揭示了不列颠哥伦比亚沿海地区picorna-like ssRNA病毒在不同时间和空间的分布规律。此外,科研人员发现热带沿海海水样品中picorna-like病毒在RNA病毒中的优势地位,以及通过标记基因的额外扩增子测序,发现RNA病毒对潜在宿主的群落组成有影响。这些数据使人们初步了解了海洋中某一特定RNA病毒群的多样性、丰富性和生态环境影响,但这种方法几乎无法提供更多遗传多样性(即其他病毒基因)的信息,而且由于病毒群的代表性不足和缺乏完整的基因数据,在基因组和功能层面的分析不足,不适用于RNA病毒的全球和综合评估。
2. 基于病毒颗粒富集的海洋RNA病毒学研究
依赖培养的方法和单基因测序技术的局限促使现代宏组学技术出现,即研究从自然环境样品中获得所有遗传物质。早期海洋病毒学研究涉及的采样地有限,并且大部分研究分析双链DNA病毒,解决这个问题需要进行大规模地采样和测序工作。
第一项以RNA病毒群落为重点的研究,是通过构建随机反转录全基因组鸟枪文库,调查了四个水生环境中未培养的RNA病毒群落,揭示了海洋中广泛存在相对较短的RNA病毒基因组(包括Picornavirales、Mulpavirales和Tolivirales病毒目中的病毒以及其他未分类和未知的病毒)。这项研究验证了RNA病毒宏基因组学作为揭开隐藏的海洋RNA病毒圈的方法的可行性。与传统方法相比,这种方法不需要复杂的分离和培养程序,在恢复病毒基因组方面具有优势。因此,早在2016年,通过宏基因组方法记录的未培养病毒的数量就已经超过了分离培养的病毒。同时,通过宏组学方法发现海洋病毒,可以有效避免采样偏差,提供RNA病毒的全球评估和生态学的视角。随后,科学家们开展了越来越多的项目来探索不同环境中的病毒,如热带海水、大西洋沿岸海域、南印度洋、加拿大北极地区、波罗的海和最深海沟,以及来自海洋生物,如硅藻、卵菌类、海洋节肢动物和真核浮游植物的病毒。RNA病毒宏基因组学对扩大我们对RNA生态特征理解做出了巨大贡献,包括海洋RNA病毒的丰度和多样性、时间动态、生物地理分布和宿主相互作用。
病毒颗粒富集方法研的进步对海洋病毒的研究有着巨大贡献。通常,基于病毒颗粒的RNA病毒学工作流程包括以下步骤:取样和细胞分层过滤、病毒颗粒富集浓缩、核酸提取和扩增、文库制备和高通量测序,最后是下游的个性化数据分析。在这些主要步骤中,可能出现偏差,必须仔细考虑以避免错误。病毒浓缩和纯化过程是为了减少样品量,方便后续步骤,经常使用的方法有切向流过滤、氯化铁絮凝、超速离心和氯化铯梯度离心方法。通过不同的筛选条件,这些方法保持了病毒的完整性,但引入了病毒类型的偏差,因为不同类型的病毒在相同筛选条件下有不同的保存水平。其次,这也可能引入外源核酸污染,这些污染可能来自样品、试剂和测序平台,应通过DNA/RNA核酸酶处理,并控制实验设计,谨慎考虑低丰度的病毒,控制并尽量减少污染,以保证结果的准确性(图2)。
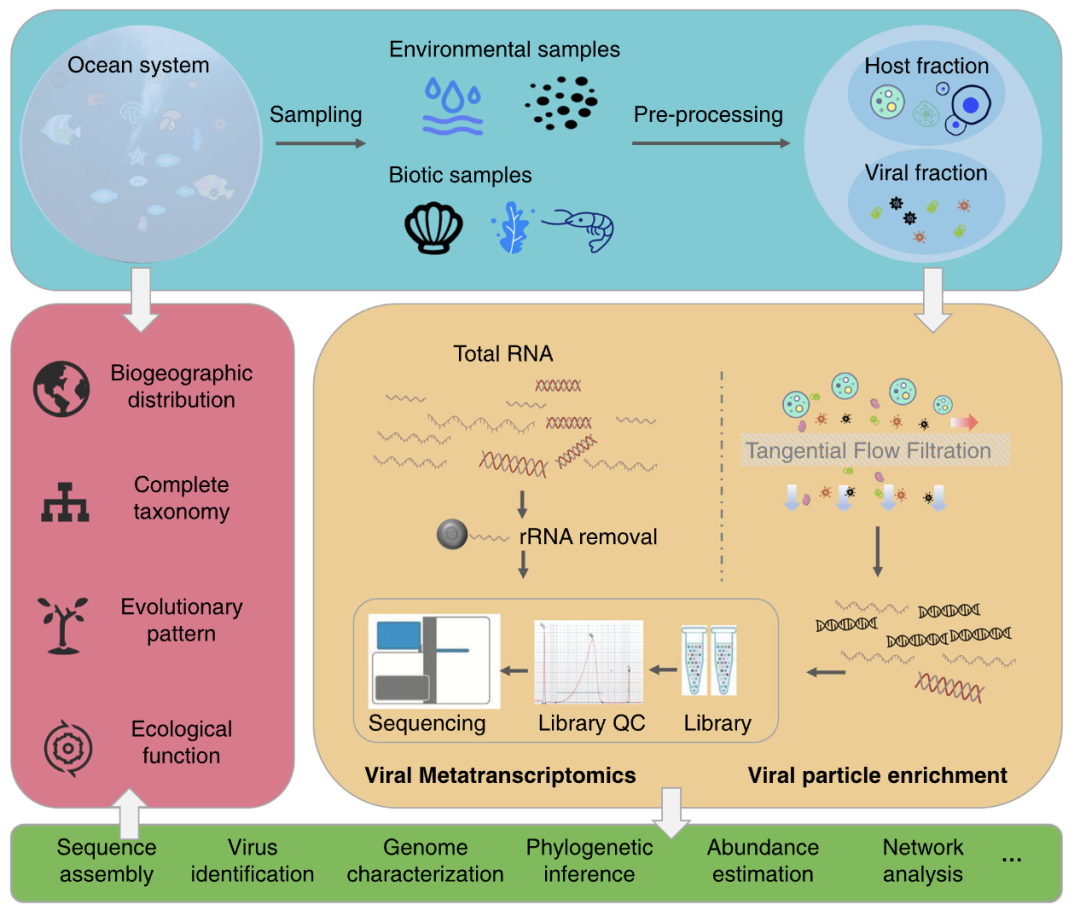
图 2. 海洋宏病毒组分析流程总结
步骤(1):实验方案设计:这一步在宏组学研究中往往不被重视,但认真执行能为后续研究提供更多更准确的信息。步骤(2):用不同方法收集病毒核酸序列信息,收集海洋RNA病毒及其宿主的信息。步骤(3):组装好病毒基因组后进行各种个性化分析。步骤(4):在病毒、宿主及环境的背景下进行详细注释,并从全球视角进行研究
3. 海洋病毒宏转录组学
数据库中不断积累的RNA序列、更好的实验方案和敏感度更高的计算工具,在过去十年里宏转录组学(在一个特定的环境中的总RNA)的应用进一步促进了RNA病毒的发现,它在提供RNA病毒(具有以RNA的基因组的病毒)以及以DNA的基因组的生物体(即细胞生命体和DNA病毒)的转录组的信息方面具有优势。一个典型的RNA病毒组分析流程包括采样(从环境或生物体组织中取样)、细胞裂解、DNA去除、RNA提取和浓缩(通过mRNA分离或rRNA去除),逆转录、测序和数据分析(图2)。
宏转录组学凭借其无偏差、广泛可用和成本效益高的特点,揭示了各种样本生境(环境样本、微生物、植物、无脊椎动物和脊椎动物)中隐藏的多样的RNA病毒。这些研究对了解海洋RNA病毒的隐藏多样性、进化情况、遗传和系统发育多样性以及生物地理分布做出了极大贡献。此外,宏转录组作为一个提供病毒及其宿主信息的多维度工具,同样被用于检测活跃的病毒感染和推断病毒与宿主以及采样地的相互作用。随着公共数据库中病毒组数据的积累,一些基于不同原理的高级生物信息学工具可以在从收集的宏基因组或宏转录组片段中识别出蛋白质,对RNA病毒进行检测和分类,这些工具包括BLAST, VirFinder, Kraken2,Vibrant, VirSorter2等。在不同模拟条件下,这些方法发现病毒的精确度、重现性和F1得分都具有较大差异。测序重叠群的长度和分类的复杂性都会影响这些工具的平均性能,因此研究人员可以根据自己的需要选择合适的工具。实际分析过程中,研究人员可以通过实验室实验(基于引物设计的RT-PCR)和多重计算确认(在已知RNA病毒的参考RdRps中考虑真实性,或将读长比对到病毒重叠群并估计丰度)来纠正可能发生的偏差。尽管以RdRp蛋白质为主的系统发育分析无法解释序列极端分歧度的情况,并且只有在核心序列明显相似的情况下才具有较高可信度。不过从基于RdRp的系统发育中得出的结论可以结合其他分类方法得出的结论综合判定(如RdRp的三维结构和聚类,其他结构域的存在和排列方式,以及全基因组特征)来确认RNA病毒的分类。此外,当有足够的生态位背景信息时,可以探讨海洋RNA病毒的生态作用和生物地理模式。
未来发展与展望
NGS和病毒组学改变了病毒的发现方法,使已知和未知的病毒序列数激增,这在一定程度上让我们理解的实际的海洋RNA病毒圈变得更复杂。与确定原核生物DNA病毒-宿主关系不同,通过基于CRISPR间隔物匹配或序列相似性的方法检测真核生物RNA病毒宿主,难度更高。即使直接对组织进行采样,也不能将推断出此物种为病毒宿主,因为病毒实际上可能感染宿主内部微生物或饮食成分。所以在未来的病毒组研究中,应鼓励旨在探索新的机制(如感染引起的序列特征共存关系)或新的技术创新的研究,来确定病毒与宿主的关联,提高方法准确性和适用性。其次,提高病毒基因组的完整度是当务之急,需要更高的测序深度、更长的测序读长和更好的组装工具。另一个难点是,很少有生物信息学软件可以用来对与已知病毒相似性低的RNA病毒进行精确分类。最后,在从环境和研究不足的生物样本中分离RNA病毒颗粒以及病毒培养学和分子验证方面仍有很大的技术缺陷,这一领域的进展将有助于人们更深入地了解RNA病毒的生物学特征,以及人类在病毒群落和/或生态系统层面对RNA病毒更广泛的研究。
通过宏组学手段发现病毒没有一成不变的方法路径,有助于揭示未被探索的RNA病毒 的未知方面。不同类型病毒的进化过程和病毒世界的广泛分类学有望通过大规模和无偏见的病毒组学得到完善。此外,阐述影响海洋RNA病毒的因素以及RNA病毒对海洋生态系统的影响同样很重要。特别是,通过整合代谢物谱和地理生化和环境特征,可以在基因组和代谢水平上确定参与碳、氮、硫和其他地球元素循环的海洋RNA病毒群的作用,从而为开发海洋病毒资源和利用RNA病毒来确定和调节陆地-海洋-大气的碳通量提供理论基础。但是,仍然需要更多的研究来支持对这些RNA病毒进行更完整的功能描述。
总 结
在这篇综述中,我们强调了通过宏组学方法在全球范围内探寻RNA病毒圈组成,了解海洋环境中隐秘而多样的RNA病毒种群,并揭示其生物地理分布和进化模式。然而,这些发现也表明目前的研究存在许多不足,特别是在特定生态系统中对目前有着繁多信息的病毒进行等级分类和功能注释。此外,研究人员还需要更先进的测序技术(更长的读长)和计算方法(尽可能完整地组装病毒基因组),以进一步提高我们对已知和尚未发现的海洋RNA病毒的认识。随着通过各种方法发现大量的RNA病毒及其与宿主和环境因子的关系,海洋RNA病毒所发挥的潜在作用,会重新定义它们在自然界的生态地位。
引文格式:
Liao, Meng-en, Xie, Yunyi, Shi, Mang, and Cui, Jie. 2022. “ Over two decades of research on the marine RNA virosphere.” iMeta e59. https://doi.org/10.1002/imt2.59
作者简介

廖蒙恩(第一作者)
● 中国科学院上海巴斯德研究所硕博连读研究生
● 主要研究兴趣为RNA病毒的多样性和进化

崔杰(通讯作者)
● 中国科学院上海巴斯德研究所研究员南、博导,病原生物信息学研究组组长
●长期从事病原发现、病毒进化、开发组学新方法等研究,重点关注:病毒生态学和病毒圈(virosphere);癌症进化;基于生物信息学的病原大数据分析
更多推荐
(▼ 点击跳转)
iMeta | 德国国家肿瘤中心顾祖光发表复杂热图(ComplexHeatmap)可视化方法
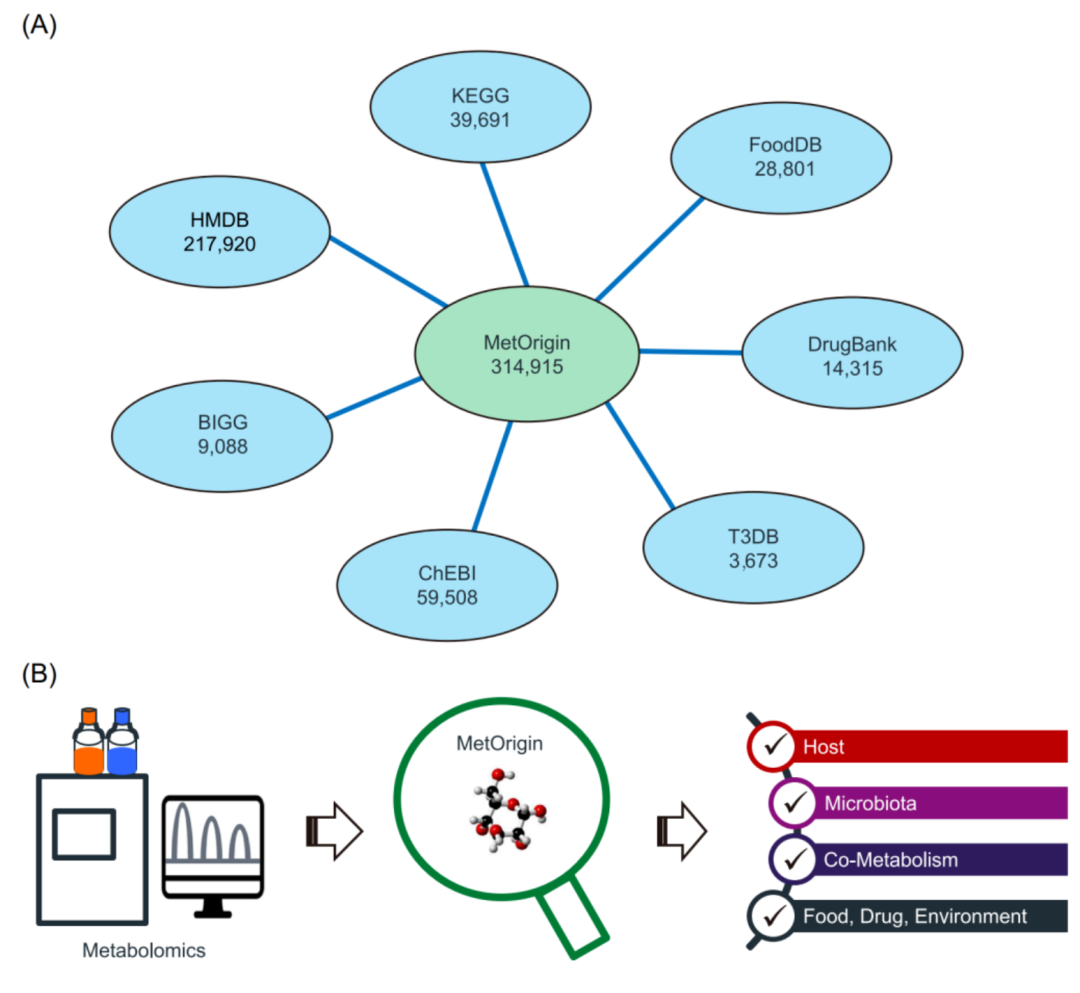
iMeta | 浙大倪艳组MetOrigin实现代谢物溯源和肠道微生物组与代谢组整合分析

第1卷第1期

第1卷第2期

第1卷第3期
期刊简介

“iMeta” 是由威立、肠菌分会和本领域数百位华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表原创研究、方法和综述以促进宏基因组学、微生物组和生物信息学发展。目标是发表前10%(IF > 15)的高影响力论文。期刊特色包括视频投稿、可重复分析、图片打磨、青年编委、前3年免出版费、50万用户的社交媒体宣传等。2022年2月正式创刊发行!
联系我们
iMeta主页:http://www.imeta.science
出版社:https://onlinelibrary.wiley.com/journal/2770596x
投稿:https://mc.manuscriptcentral.com/imeta
邮箱:office@imeta.science
微信公众号
iMeta
责任编辑
微微
往期精品(点击图片直达文字对应教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|




























后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集




















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









