







时间:2020
刊物:Nature Genetics
题目:神经突生成相关基因的突变诱发脑瘫发生风险
作者:Sheng Chih Jin等,耶鲁大学医学院遗传学系
链接:https://www.nature.com/articles/s41588-020-0695-1
01 | 摘要
除了常见的环境因素外,基因组因素也可能导致脑瘫。我们对250个亲本后代(parent–offspring trios)进行了全外显子组测序,观察到脑瘫病例中具有有害(damaging)新生突变(de novo mutations)的富集。(我们发现)8个基因有多个有害新生突变;其中,两个基因(TUBA1A和CTNNB1)具有全基因组意义(genome-wide significance)。我们发现了两个新的单基因致病因素(monogenic etiologies):FBXO31和RHOB,并阐述了RHOB的突变增强了激活态Rho效应结合(enhances active-state Rho effector binding),而FBXO31的突变降低了周期蛋白D(cyclin D)的水平。(此外还发现)脑瘫风险候选基因与神经发育障碍基因(存在)重叠。通过网络分析发现了以下通路的富集:Rho GTPase,细胞外基质,局灶粘附(focal adhesion)和细胞骨架。在果蝇反向遗传筛选中,富集通路中的脑瘫风险基因被证明可以调节神经运动功能。我们估算14%的病例可归因于过量的有害(damaging)新生或隐性变异(de novo or recessive variants)。这些发现为(通过)遗传介导的(genetically mediated)脑瘫早期神经元连接失调,提供了证据。
脑瘫(cerebral palsy),即脑性瘫痪,是指婴儿出生前到出生后一个月内脑发育早期,由于多种原因导致的非进行性脑损伤综合征。主要表现为中枢性运动障碍以及姿势异常,还可伴有智力低下、癫痫、感知觉障碍、语言障碍及精神行为异常等,是引起小儿机体运动残疾的主要疾病之一。
脑瘫病因:脑瘫的高危因素主要发生在缺氧缺血性脑病、早产、高胆红素血症、颅内出血等一项或多项因素的新生儿,其中部分可能发展为脑瘫。与脑瘫相关的风险因素并不会(直接)导致这种疾病,但会增加儿童出生时患有这种疾病的机会。脑瘫不是遗传性的。(Risk factors associated with CP do not cause the disorder but can increasea child's chance of being born with the disorder. CP is not hereditary.)
TUBA1A:该基因编码α微管蛋白,与小鼠和大鼠Tuba1基因高度相似。Northern blot研究表明,该基因主要在形态分化的神经细胞中表达。该基因突变导致3型无脑畸形(LIS3),这是一种以小头症、智力残疾和由神经元迁移缺陷引起的早发型癫痫为特征的神经疾病。其相关途径包括细胞器的生物发生和维持以及发育的Slit-Robo信号通路。与该基因相关的GO注释包括GTP结合和结构分子活性。
CTNNB1:该基因编码的蛋白质是组成粘附连接(adherens junctions,AJs)的蛋白质复合物的一部分。AJs通过调节细胞生长和细胞间黏附而形成和维持上皮细胞层所必需的。编码的蛋白质还锚定肌动蛋白细胞骨架,并可能负责传递接触抑制信号,导致细胞停止分裂。该蛋白可与APC基因的产物结合。该基因在结肠腺瘤性息肉病中发生了突变。该基因突变是结直肠癌(CRC)、毛基质瘤(PTR)、髓母细胞瘤(MDB)和卵巢癌的一个原因。与CTNNB1相关的疾病包括:毛基质瘤和神经发育障碍伴痉挛性双瘫和视觉缺陷。相关通路:胃癌网络2和TNFR1通路。GO注释包括:DNA结合转录因子活性和结合。该基因是经典Wnt信号通路的下游关键成分。参与细胞粘附的调节,作为E-cadherin:catenin粘附复合物的组成部分(通过相似性)。作为中心体内聚的负调控因子。参与胰岛素内化的CDK2/PTPN6/CTNNB1/CEACAM1通路。通过下调DAPK2,阻断恶性肾和肠上皮细胞的anoikis,促进其锚定非依赖性生长。通过维持细胞周期内的交感神经母细胞促进神经发生。通过与SOX9相互作用参与软骨细胞分化:SOX9结合与CTNNB1内TCF/LEF结合位点竞争,从而抑制Wnt信号通路。
FBXO31:是F-box家族的一员,是SCF泛素化复合物的一个组成部分。它被认为是结合和招募底物进行泛素化和降解。这种蛋白可能在调节细胞周期以及树突生长和神经元迁移中发挥作用。与FBXO31相关的疾病包括:智力发育迟缓和常染色体隐性非综合征性智力残疾。其相关途径包括I类MHC介导的抗原加工和呈递和先天免疫系统。与该基因相关的GO注释包括:细胞周期蛋白结。特异性识别磷酸化的cyclin-D1 (CCND1),促进其泛素化和被蛋白酶体降解,导致G1阻滞,亦可能作为肿瘤抑制因子。
RHOB:与RHOB(Ras Homolog Family Member B)相关的疾病包括肺癌和难辨梭菌性结肠炎(Clostridium Difficile Colitis)。其相关通路有RhoGDI通路和PAK通路。与该基因相关的GO注释包括GTP结合和GDP结合。该基因介导DNA损伤后肿瘤转化细胞的凋亡。不是发育所必需的,但在转化细胞中影响细胞粘附和生长因子信号。在肿瘤发生中起负作用,因为缺失会导致肿瘤形成。参与细胞内蛋白质运输的一些蛋白质。作为一个微管依赖的信号,在细胞周期和胞质分裂期间肌凝蛋白收缩环的形成。在乳腺癌细胞中被基因毒性应激诱导的细胞死亡所需。
02 | 前言
脑瘫(CP)是影响运动功能的主要神经发育障碍(neurodevelopmental disorder, NDD),世界范围内每1000名儿童中约有2~3名受到影响。运动障碍(痉挛性,肌张力障碍,舞蹈手足徐动症和/或共济失调)发生在生命的最初几年,是大脑发育中断后的表征。从历史上看,尽管利特尔和奥斯勒(Little and Osler)认为CP主要是围产期缺氧造成的,但弗洛伊德(Freud)对这一说法提出了异议。直到今天,关于CP起源的争论仍在继续,特别是在个别案例中,具有广泛的医学和法律影响。
正如其他NDDs,如自闭症谱系障碍(autism spectrum disorders, ASDs)和智力残疾(intellectual disability, ID),虽然一些环境因素,包括早产、感染、缺氧缺血和产前和围产期中风,是CP风险的主要因素,但没有单一的致病因素与CP有关。然而,多达40%的CP病例可能没有一个容易识别的病因,定义为隐源性或特发性(cryptogenic or idiopathic)CP。基于登记的数据显示,21%~40%的CP病例有相关的先天性异常,其中许多病例涉及基因组改变。在一个瑞典西部(人群)队列CP病因学的概率模型支持下,预估CP的遗传率/遗传可能性(heritability)为40%,与ASD的38%~58%的遗传率相当。
迄今为止,已有5项研究分析了CP病例的基因组拷贝数变异(copy number variations, CNVs),在10.31%的病例中发现了预测的有害CNVs。此前已有三项全外显子组测序(Whole Exome Sequencing,WES)研究在CP病例中进行。迄今为止,最大的一项研究报告称,在98个未选择类型CP(unselected forms of CP)的亲本后代(parent–offspring trios)中,约有14%的突变体是有害(deleterious)的。这些研究表明CP有潜在的重要遗传风险,但由于缺乏可用的、充分的对照(组)限制了的统计学推断的进行,且未对新的候选基因变异进行功能验证。我们试图在本研究中解决这些局限性。
03 | 结果
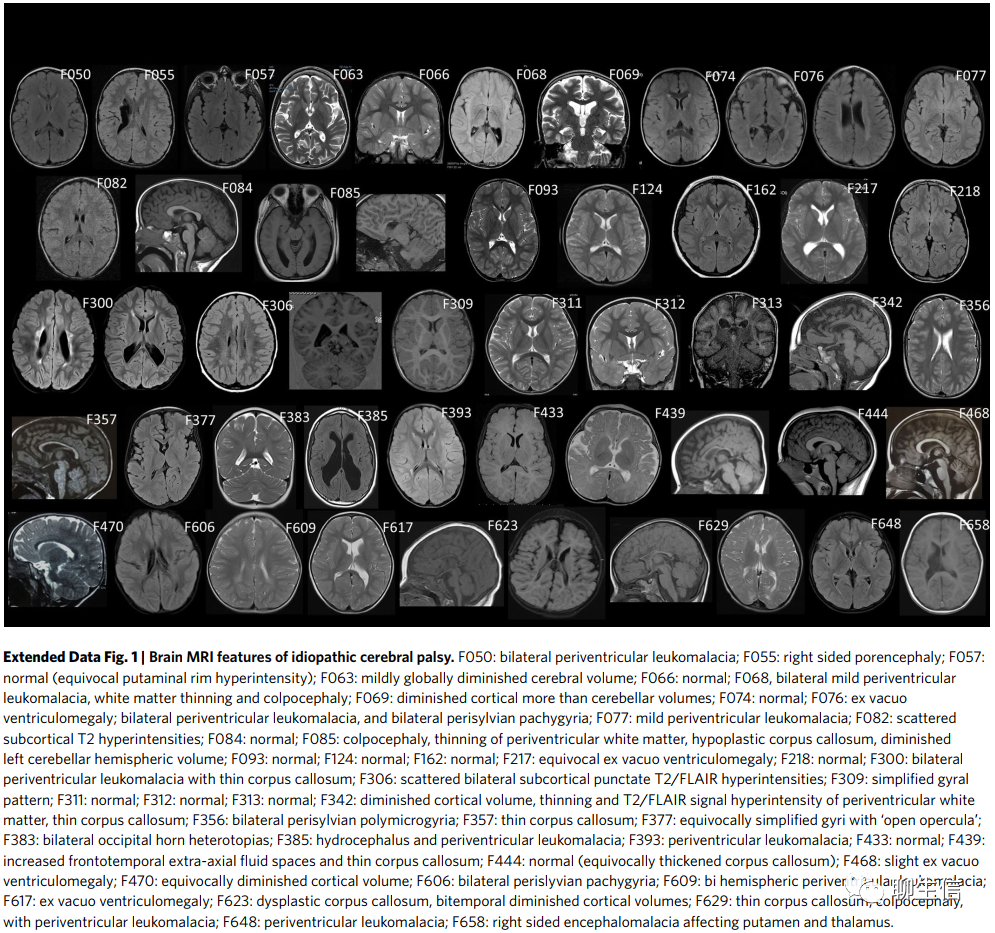
脑瘫队列人群特征及外显子测序(CP cohort characteristics and WES)。根据当地的伦理要求,在获得书面知情同意后,我们对250例CP三人组家系(trios)进行了WES(全外显子组测序),其中包括之前报道的91例,以及来自美国、中国和澳大利亚的159例。病例由临床专家根据国际共识标准进行诊断(Supplementary Table 1 and Supplementary Dataset 1),故CP被定义为一种运动和(或)姿势损害运动功能的非进行性(non-progressive)发育障碍。病例在两岁时出现症状。因此,这个操作性定义(operational definition)排除了进行性神经疾病,如神经退行性疾病。无已知染色体异常或非整倍体,无临床或分子诊断综合征(如Rett综合征、Angelman综合征),无致病性微缺失或微重复综合征,无线粒体疾病或创伤性脑损伤。详细的患者表型可见Supplementary Note,具有代表性的神经影像学发现见扩展数据图1(Extended Data Fig. 1),具有代表性个体的运动障碍表型的视频可在补充视频(43个视频可通过https://figshare.com/s/a4f914ab77958ab3e4b6获得)和补充照片(https://figshare.com/s/0f200402e51de5875390)中找到。
视频截图:

照片截图:

在250个三人组家庭队列中,157例(62.8%)被归类为特发性(idiopathic)(原因不明),84例(33.6%)有已知的与CP相关的环境损害(包括妊娠<32周的早产,围产期缺氧缺血(由临床医生定义),缺血性/出血性中风和/或感染),其余9个三人组(3.6%)不能被分配到任何一种类别(“unclassified”; Supplementary Table 1)。
如之前的文献中所述(的方法),进行WES检测(见Supplementary Table 2:外显子组数据表)。自闭症患者的未受影响的兄弟姐妹和他们的未受影响的父母(共1789名,由Simons Simplex Collection提供)组成的对照组作为平行对照进行分析(Control trios consisting of 1,789 unaffected siblings of autism cases and their unaffected parents from the Simons Simplex Collection were analyzed in parallel)。使用BWA-MEM(软件)比对测序reads,使用GATK Best Practices检测变异(call variants)。使用MetaSVM和Combined Annotation Dependent Depletion (CADD v1.3)算法预测有害错义突变(deleteriousness,本文定义为D-Mis,MetaSVM-deleterious或CADD>20)。推断的功能缺失(loss-of-function,LoF)变异包括:终止突变(stop-gain)、终止丢失(stop-loss)、移码插入/缺失(frameshift insertions/deletions)、经典可变剪接位点(canonical splice sites)和起始丢失(start-loss)。LoF和D-Mis突变均被(我们)认为是有害的(“damaging”)。使用TrioDeNovo软件进行新发突变(De novo mutation,DNMs)检测。使用一代测序验证感兴趣(目标)基因(genes of interest)的突变。
Combined Annotation Dependent Depletion(CADD):CADD是对人类基因组中单核苷酸变异以及插入/删除变异的危害性进行评分的工具/网站。虽然有许多不同的注释和评分工具,但大多数注释倾向于利用单一的信息类型(例如:保守性)和/或在适用范围上受限(例如:错义突变)。因此,需要一个广泛适用的度量标准,客观地衡量和集成各种信息。CADD是一种框架,通过对比自然选择中幸存的变体和模拟突变,将多个注释集成到一个数据集中。C-scores 与等位基因多样性、编码和非编码变异的致病性、实验测量的调控效应以及个体基因组序列中的Top致病变异(highly rank causal variants)密切相关。最后,全基因组关联研究(GWAS)中复杂性状相关变异的C-scores 显著高于匹配的对照,并与研究样本量相关,这可能反映了更大的GWAS准确性的提高。CADD可以在广泛的功能类别、效应值和遗传结构中,定量地对功能性、有害性和致病性变异进行优先级排序,亦可在科研和临床环境中对致病变异进行优先性排序。
CP队列显著富集了有害新发突变(Damaging DNMs are significantly enriched in the CP cohort)。 我们首先在队列中评估DNMs(de novo mutations)对CP的贡献的水平。 疾病组和对照组中观察到的DNMs数量近似于泊松分布(Extended Data Fig. 2),表明DNMs是独立的概率事件(independent probabilistic events)。
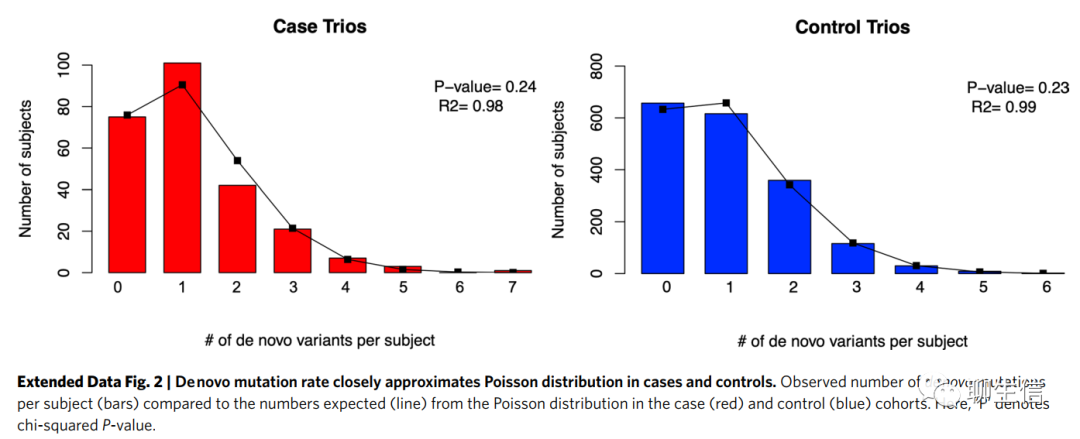
我们发现在CP组中,可以显著富集到有害新发突变(damaging DNMs;enrichment=1.22;P=7.4×10-3;Table 1),而对于不耐受(intolerant)LoF变异,CP组的富集更加明显(pLI score≥0.9 in gnomAD v2.1.1)(enrichment=1.78;P=1.2×10-5)。在对照中没有发现任何突变类别的显著富集(Table 1)。
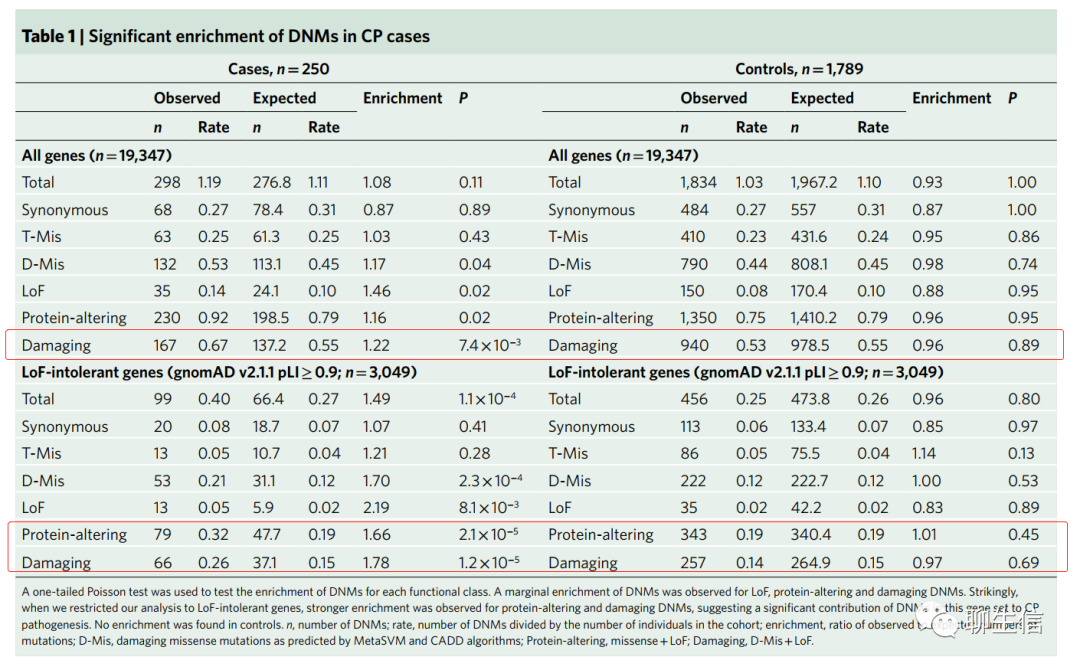
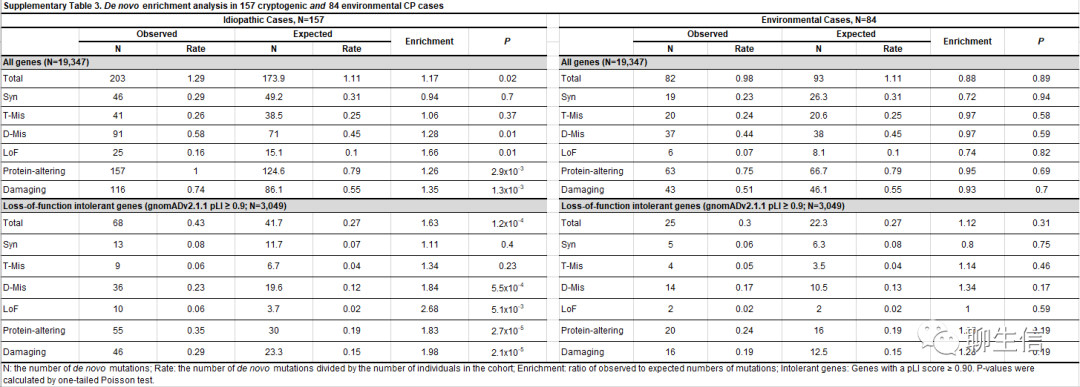
当我们考虑确定性差异(ascertainment differential)(即:观察到的有害DNMs数量与预期有害DNMs数量进行比较,除以队列中三人组trios的数量),我们队列中11.9%的CP病例可归因于过量的有害新发突变(excess of damaging DNMs)。当按CP亚型对病例进行分层(stratifying)时,我们发现与环境性病例(environmental cases)(enrichment=1.28; P=0.19; Supplementary Table 3)相比,特发性有害DNMs富集更大(enrichment=1.98; P=2.1×10-5),暗示特发性病例具有更高的有害DNMs负荷(burden)。
Supplementary Table 3
反复出现的有害新发突变涉及了已知的和新的CP基因(Recurrent damaging DNMs implicate both known and novel CP genes)。接下来,通过从头机制(de novo mechanism)我们着重考虑了在我们的CP队列中反复涉及到的个别基因(Supplementary Dataset 2)。我们鉴定了8个含有≥2个有害DNMs的基因(宋红卫:一个基因内部有时可检测到多个有害突变位点)。TUBA1A(P=4.8×10−8)和CTNNB1(P=9.8×10−10)在全基因组显著性方面超过了Bonferroni校正阈值(Bonferroni correction cutoffs)(Table 2和Supplementary Table 4)。我们观察到这些基因中的蛋白损伤(protein-damaging)DNMs强烈暗示这些基因是真正的(bona fide)CP相关基因。在这8个基因中,ATL1、CTNNB1、SPAST和TUBA1A此前已被证实与人类CP表型相关。我们还在RHOB和FBXO31这两个基因中发现了相同但独立产生的有害新发突变(damaging DNMs)。
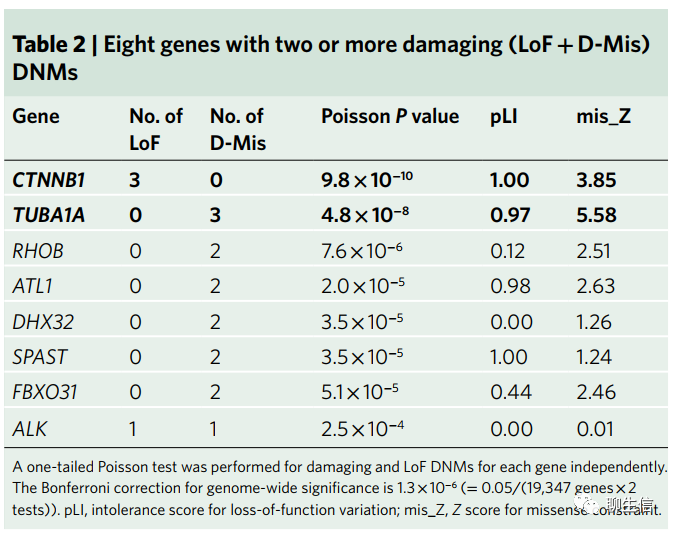
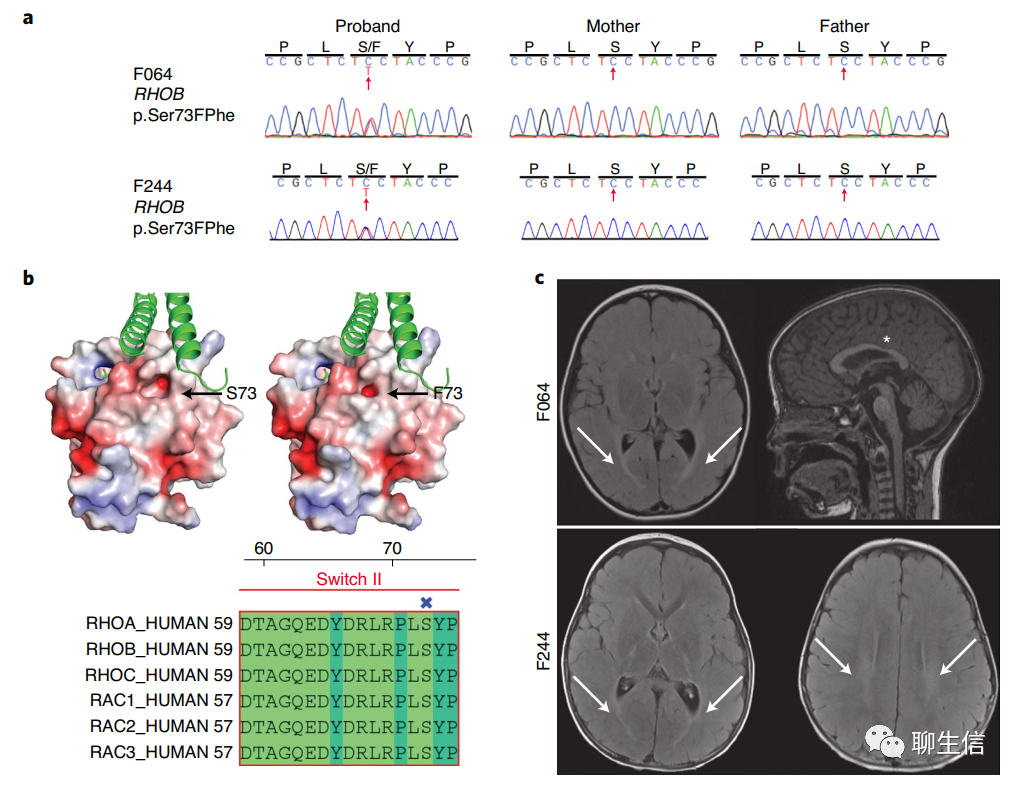
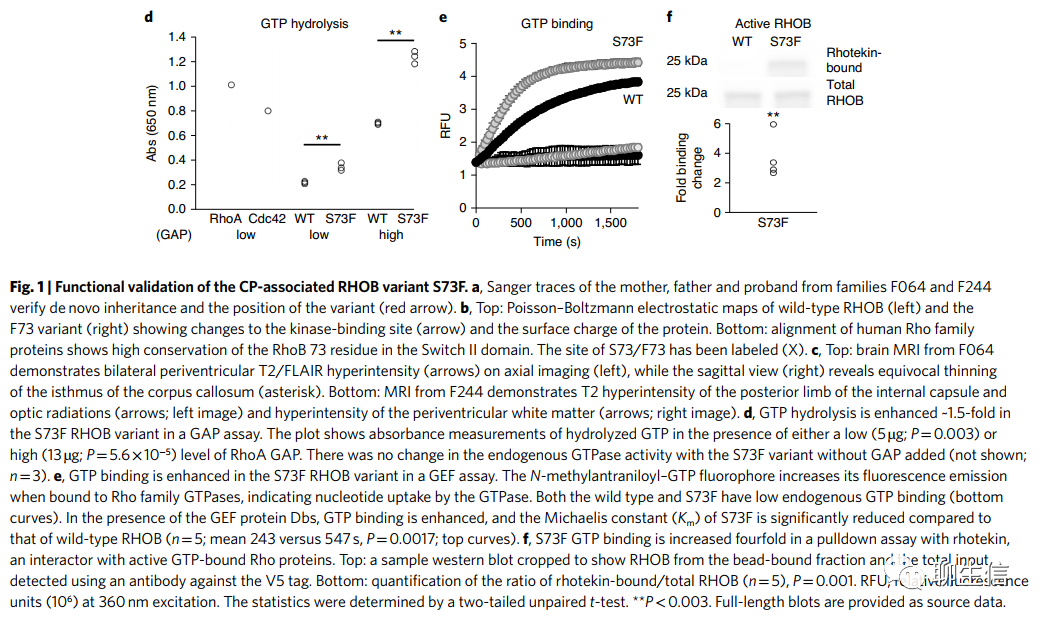
RHOB和FBXO31具有相同的功能获得性新发突变(gain-of-function DNMs)。RHOB编码Rho GTPase,在两例不相关的痉挛性张力性脑瘫病例中含有两个相同的DNMs(编码p.Ser73Phe;Fig. 1a和Supplementary Table 4),代表一个不太可能的偶发事件(P=1.6×10-3; Supplementary Note)。Ser73预测可被磷酸化(0.997 by NetPhos 3.1),并位于Switch II结构域的一个保守位置(Rho蛋白激酶与Rho-和Rac-相关蛋白相关联的位置)(Fig.1b)。比较RHOB的野生型和p.Ser73Phe的结构模型可以发现,结合位点的形状和蛋白质的表面电荷都发生了变化(Fig. 1b)。两名患者均具有显著的一致性表型,包括磁共振成像(MRI)上T2白质高信号(a hyperintense T2 white matter signal)(脑室周围白质软化)、痉挛性肌张力异常双瘫、表达性语言障碍和主动脉弓异常(Fig. 1c,Supplementary Table 4和Supplementary Videos F064 and F244)。已知RHOB可控制树突棘的生长,但此前没有发现与人类疾病有关。生化分析表明,该变体对GTPase激活蛋白(GAPs)和GDP交换因子(GEFs;Fig. 1d,e)的增强反应,最终导致活性状态下与Rho效应rhotekin的结合增强(enhanced binding in the active state to the Rho effector rhotekin)(Fig. 1f)。
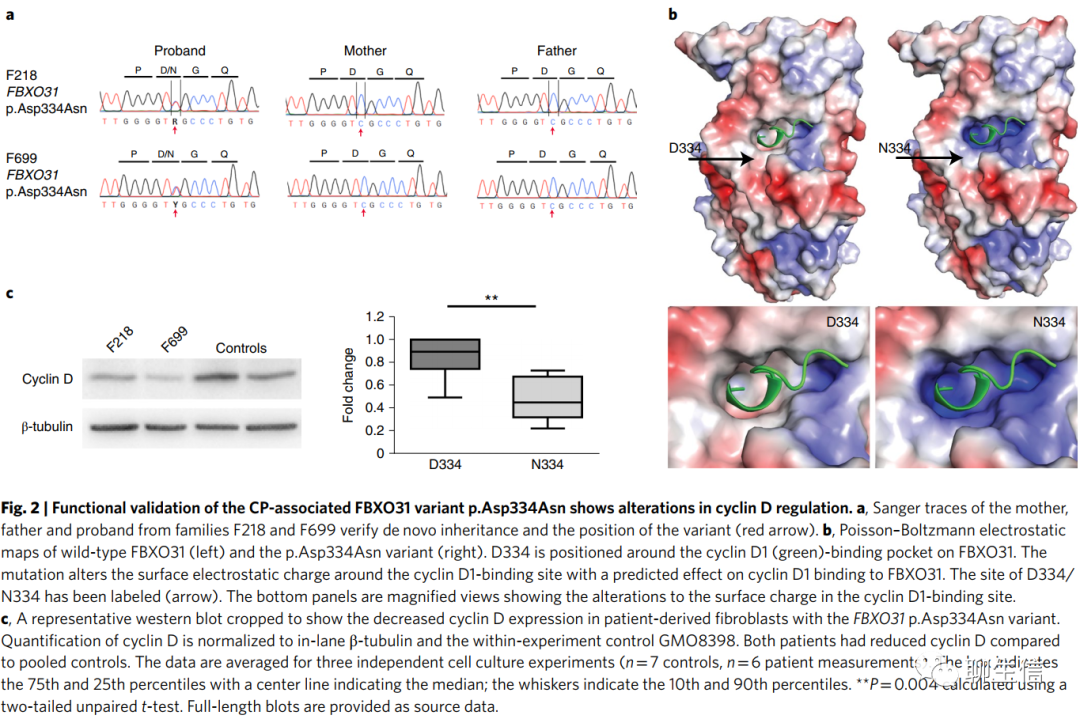
我们还确定了两个不相关的病例但包含相同的位于FBXO31中的DNM(编码p.Asp334Asn;Fig .2a和补充Table 4),它编码唯F-box蛋白31。一个FBXO31/SKP1/Cullin1复合物通过标记蛋白酶体降解的细胞周期蛋白D来控制蛋白质丰度。Asp334是一种在FBXO31上的胶袋内的保守残基(图2b),它被认为介导了与cyclin D1的氢键。众所周知,FBXO31控制轴突生长,对树突生长和神经元迁移至关重要。在发育中的大脑中。FBXO31的p.Asp334Asn突变影响细胞周期蛋白D相互作用位点(图2b),导致对细胞周期蛋白D降解功能的明显增强(图2c)。FBXO31的纯合子截短突变以前曾被报道与ID(智力障碍)有关(MIM 615979)。我们队列中的两名患者均表现为痉挛性双瘫性CP(补充表4和补充视频F218和F699), ID(智力障碍),表达性语言障碍和注意力缺陷/多动障碍。F218(病人)有肠道旋转不良和便秘,MRI(核磁共振)显示腭裂,斜视和正常的大脑形态,F699 (病人)MRI显示斜视、严重便秘、脑室扩张、胼胝体薄。因此,这个FBXO31的DNM(新发突变)导致了一种与前面描述的常染色体隐性截短突变相关的非综合征性ID(智力障碍)表型所不同的表型。
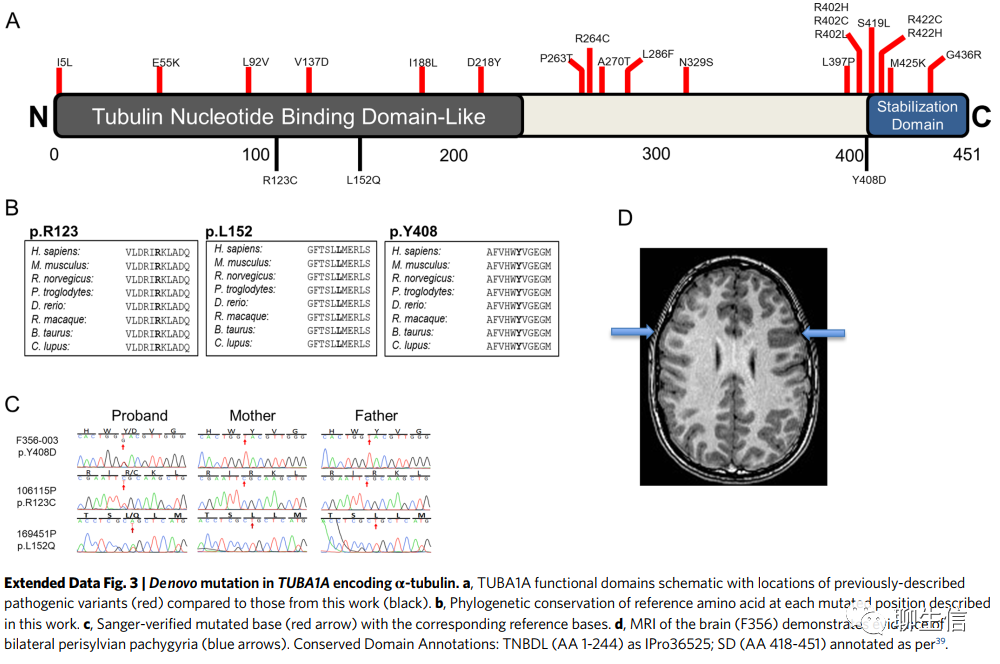
前文中DNM涉及的基因:TUBA1A, CTNNB1, ATL1和SPAST(DNMs in previously implicated genes TUBA1A, CTNNB1, ATL1 and SPAST)。TUBA1A编码微管相关蛋白α-微管蛋白,在三个不相关的先证者(probands)中含有三个有害DNMs(p.Arg123Cys,p.Leu152Gln和p.Tyr408Asp;Supplementary Table 4),其中两个已被报道。p.Arg123Cys和p.Leu152Gln都映射到微管蛋白核苷酸结合域样结构域(tubulin nucleotide-binding domain-like domain),p.Tyr408Asp映射到羧基末端稳定域(carboxy-terminal stabilization domain,Extended Data Fig. 3)。我们的患者表现出这一频谱的MRI结果(Extended Data Fig. 3)。在临床上,我们的病例表现出下肢痉挛,3例中有2例出现共同的ID(智力障碍)。
先证者(probands):指在对某个遗传性状进行家系调查时,其家系中第一个被确诊的那个人。在遗传病的家系调查中最初在医院受到检查的患者就是先证者,通常每一家系中有一个人是先证者,但在检查地区内的全体人员的时候,则所有患者都是先证者。在谱系图上通常用箭头或手指图形来表示先证者。
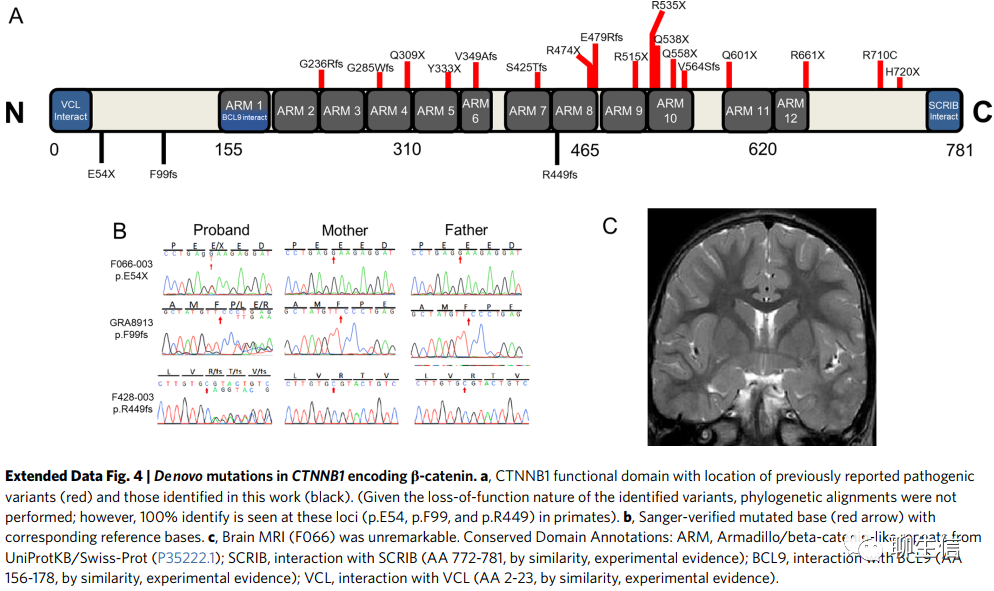
CTNNB1编码β-catenin,含有3个LoF DNMs(功能缺失新发突变,编码p.Glu54*(宋红卫:*号表示终止密码子),p.Phe99PhefsTer5和p.Arg449GlnfsTer24;补充表4),发生在三个不相关的先证者中,其中一个先前已被报道。p.Glu54*和p.Phe99fs位于氨基末端结构域,预计会导致无义(突变)介导的(蛋白)降解(nonsense-mediated decay),而p.Arg449fs位于中央犰狳重复域(central armadillo repeat domain),CTNNB1的常染色体显性种系失活突变(Autosomal dominant germline inactivating mutations)与渗出性玻璃体视网膜病变(MIM 617572)和伴有痉挛双瘫和视觉缺陷的NDD(neurodevelopmental disorder,NDD,神经发育障碍) (MIM 615075)有关。我们所有的病人都表现出痉挛,智力障碍,行为问题和语言障碍。我们还发现三分之二的病人有肌张力障碍和小头畸形。其中一名患者可能有双侧额叶回厚,但其他患者明显没有大脑检查结果(Extended Data Fig. 4)。我们发现三分之二的患者有斜视,但没有其他视觉缺陷。
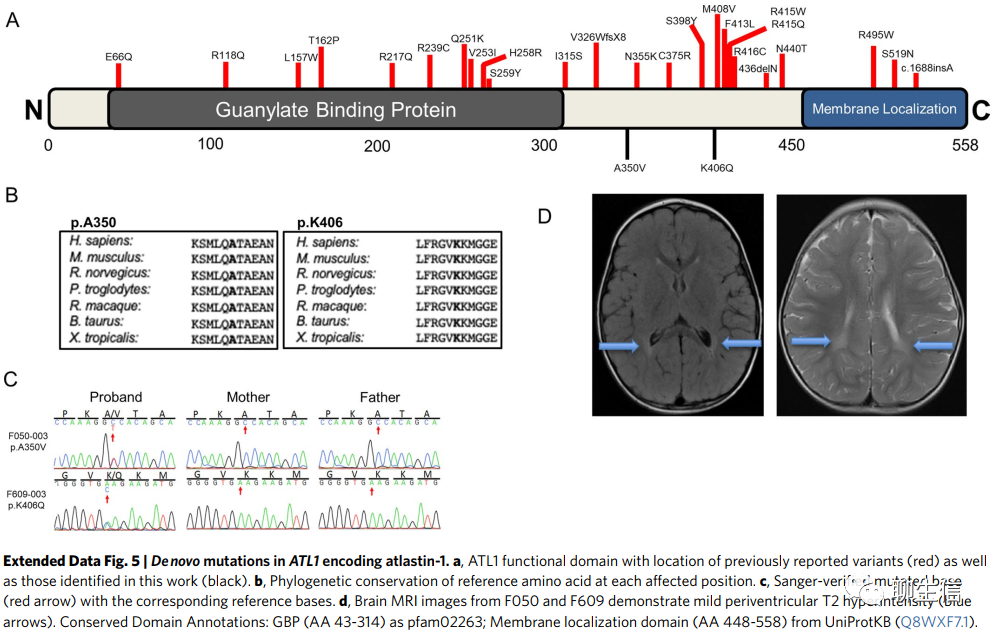
ATL1编码atlastin-1蛋白,它对神经元管状内质网的形成和轴突的伸长至关重要。在我们的队列中,ATL1携带两种有害DNMs(编码p.Ala350Val和p.Lys406Gln),位于GBP结构域;常染色体显性种系突变与神经病变1D型(MIM 613708)和痉挛性截瘫3A型(MIM 182600)有关。我们的患者表现为痉挛性和肌张力障碍,大脑表现为T2高信号和双半球脑室周围白质软化(Extended Data Fig. 5)。在最后一次随访时(患者年龄10岁和29个月)没有表型进展的迹象。
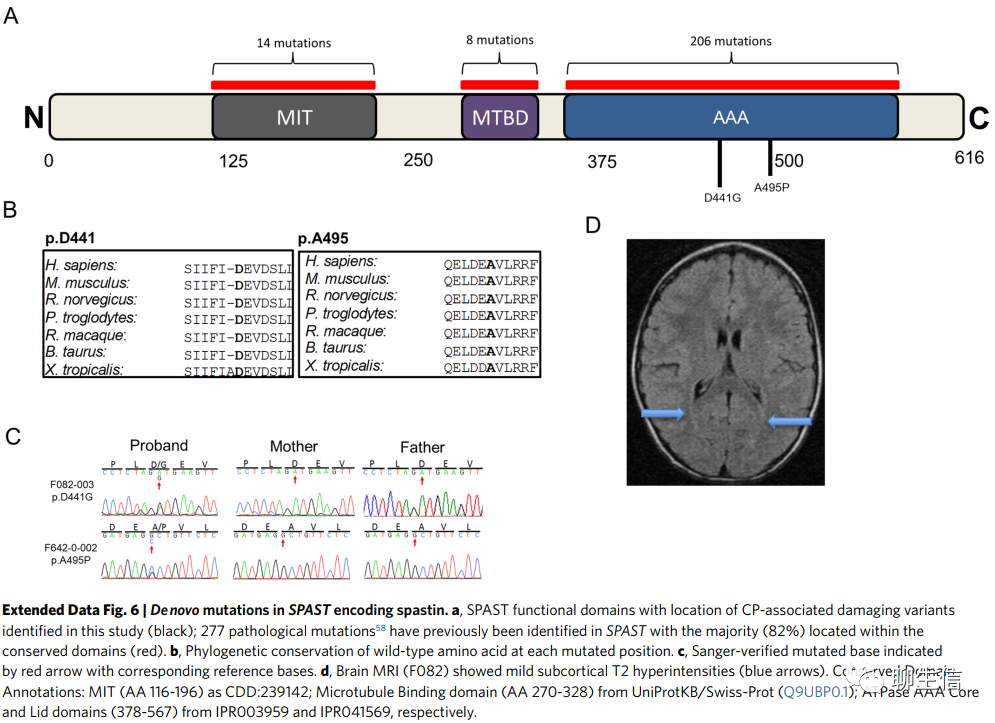
SPAST编码spastin蛋白,含有两个有害DNMs(编码p.Asp441Gly和p.Ala495Pro)。这两种突变都发生在AAA结构域的保守位置,这对ATPase活性的调控至关重要(扩展数据图6)。SPAST常染色体显性种系突变与痉挛性截瘫有关4(参考文献51;MIM 182601)。p.Asp441Gly已被报道与遗传性痉挛性截瘫(hereditary spastic paraplegia,HSP)有关。我们的患者表现为痉挛,其中一名患者还表现为肌张力障碍,其中一名患者存在散在皮层下的T2高信号(MRI),另一名患者无脑部扫描(brain findings)(扩展数据图6)。(患者)没有表型进展的迹象(患者年龄分别为21岁和40个月)。
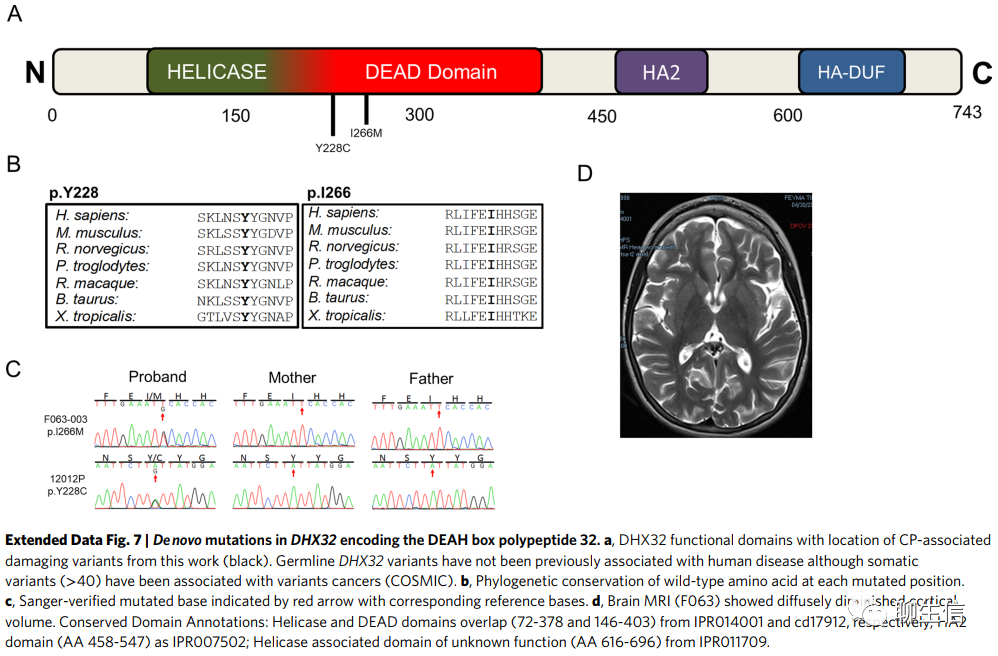
DHX32和ALK中的DNMs(DNMs in DHX32 and ALK)。DHX32编码假定的pre-mRNA剪接因子ATP依赖的RNA解旋酶DHX32,含有两个有害DNMs(编码p.Tyr228Cys和p.Ile266Met)。p.Tyr228Cys属于解旋酶ATP结合区域,此结合域是ATP结合、水解和核酸底物结合所必需的(扩展数据图7)。DHX32突变以前未被发现与人类疾病相关。我们的两个病人都表现出智力障碍,一个表现为痉挛性双瘫,另一个表现为全身性肌张力障碍。脑部检查结果(Brain findings)包括脑室周围白质软化和轻度的脑容量减少(扩展数据图7)。
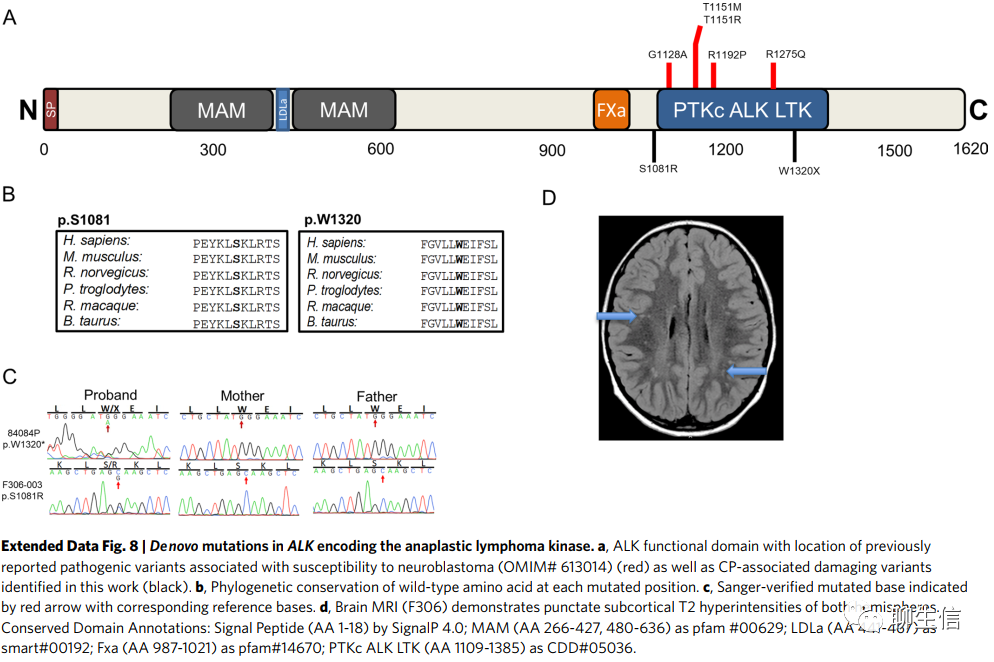
ALK,编码ALK受体酪氨酸激酶,含有一个有害DNM(编码p.Ser1081Arg)和一个终止突变DNM(编码p.Trp1320*;补充表4)。p.Trp1320*位于酪氨酸激酶结构域,而p.Ser1081Arg位于近膜结构域的上游(扩展数据图8)。ALK的种系和体细胞激活突变此前已被证实与神经母细胞瘤(MIM 613014)有关。1例患者表现为痉挛性双瘫伴轻度震颤,(检测到)散在的皮层下高信号(扩展数据图8)和心房间隔缺损。另一名患者患有痉挛性肌张力障碍双瘫,脑白质异常和癫痫。两名患者均未发现神经母细胞瘤。
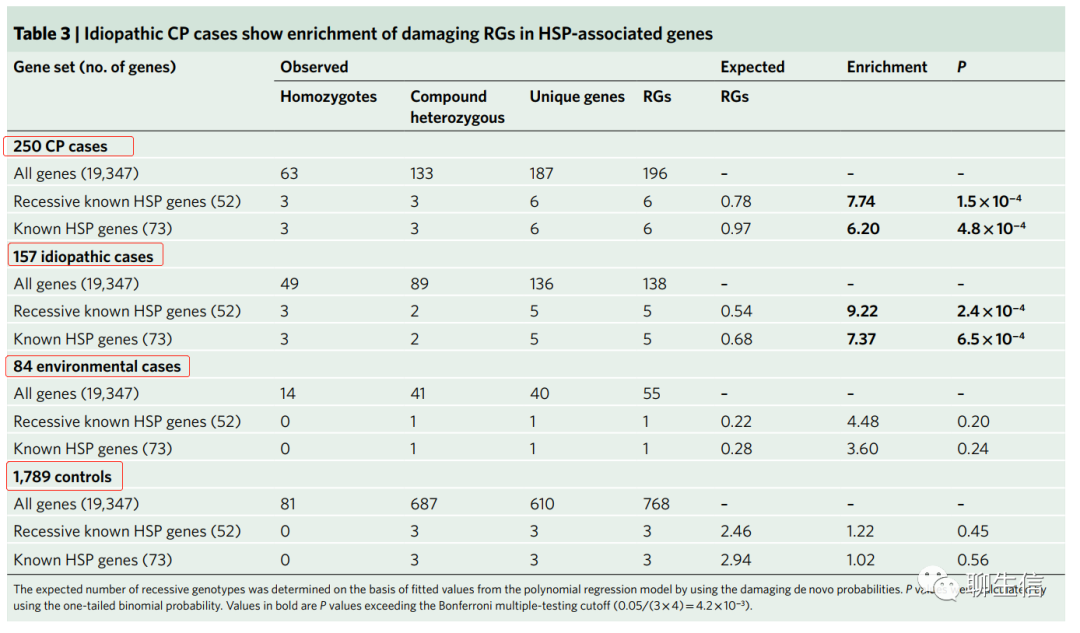
HSP(遗传性痉挛性截瘫)相关基因的隐性基因型富集(Enriched recessive genotypes in genes associated with HSP)。我们进行了单侧二项检验,并结合多项式模型来评估CP队列中每个基因的隐性基因型(recessive genotypes,RGs)负荷(补充数据集3)。在符合全基因组意义的队列中,我们没有观察到有害RGs的富集(补充表6)。我们注意到之前与HSP相关的几个基因中存在双等位基因有害变异。临床上,HSP与CP的区别在于HSP具有进行性、神经退行性以及在许多病例中发病较晚(通常已成年)。
我们仔细地重新评估了这些病例的临床表型,并发现从确诊时起没有进展的证据。有趣的是,早发伴长期临床稳定性在HSP相关基因突变患者亚群中已被确定为内表型(endophenotype)。例如,患有SPAST错义突变的患者(就像我们的病例一样)可能在蹒跚学步的时期发病,临床稳定性与CP表型一致。相反,SPAST截短突变往往会随着时间的推移被翻译和积累,推测会导致更晚的发病和神经退行性进程。此外,SPAST和ATL1在发育性神经发生中发挥重要作用,表明它们在神经元发育中的重要作用。
我们观察到6个有害RGs(隐性基因型负荷,AMPD2, AP4M1, AP5Z1, FARS2, NT5C2和SPG11;补充表7),存在于先前与隐性HSP相关的基因中(Supplementary Dataset 4;enrichment=7.74;one-tailed binomial P=1.5×10-4;Table 3)。通过差异检查(ascertainment differential),我们队列中约2.1%的CP病例可以被过量的RGs所解释。已知的HSP相关基因的RGs,主要在特发性病例中显著富集(idiopathic enrichment=9.22; one-tailed binomial P=2.4×10-4 versus environmental enrichment=4.48; one-tailed binomial P=0.20; Table 3)。
罕见的X连锁半合子变异(hemizygous variants)没有基因被富集到(No gene was enriched for rare X-linked hemizygous variants)。男性是患CP的一个危险因素。因此,我们比较了154名男性CP先证者和对照组男性罕见半合子变异(次等位基因频率,minor allele frequency(MAF)≤5.0×10-5)。没有一个基因超过了Bonferroni校正阈值(补充表8),这表明目前的研究在评估半合子负荷方面的统计能力不足。
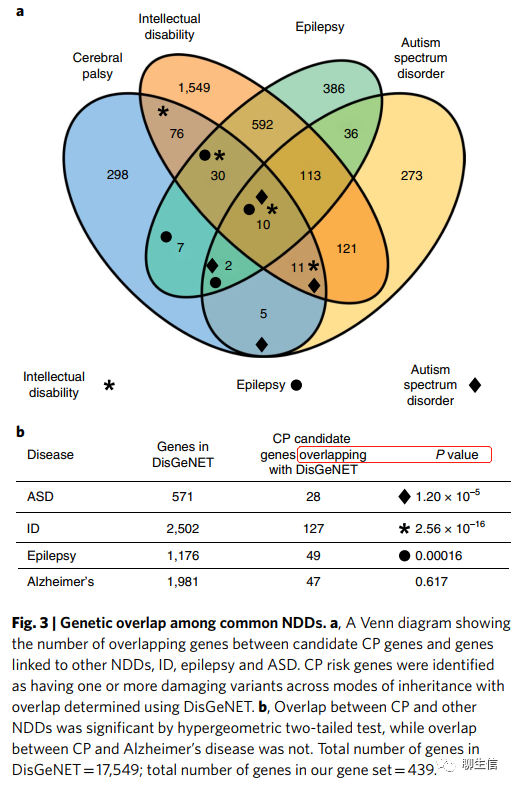
CP与其他NDDs的临床和遗传共性(Clinical and genetic overlap of CP with other NDDs)。临床上,NDDs经常具有共现性(co-occur)。(例如)在CP病人中, 约45%CP患者并发存在智力障碍, 约40%也存在癫痫,且约7% 患有ASD(自闭症)。因而我们试图确定已知的NDD风险基因与我们CP队列中的罕见有害变异基因(新发、X连锁隐性或常染色体隐性片段的假定CP基因风险;n=439,补充数据集6-15)之间的重叠程度。我们使用疾病与基因的网络工具(disease–gene network tool)DisGeNET进行分析,该工具从文献和数据库(包括ClinVar、ClinGen和UniProt)中识别基因和疾病之间的关联。我们发现CP候选基因列表和主要的NDDs之间存在大量的遗传共性(CP versus ID,enrichment=2.0,P=2.56×10−16;CP versus epilepsy, enrichment=1.7, P=1.6×10−4;CP versus ASD,enrichment=2.0,P=1.2×10−5;hypergeometric two-tailed test;Fig. 3a)。相比之下,当我们检查与神经退行性疾病(阿尔茨海默病)的重叠时,没有发现富集(图3b)。总共有28.9%的CP风险基因与智力障碍相关基因重叠,癫痫为11.1%,ASD为6.3%。我们的数据表明,CP与其他遗传性NDDs有显著的遗传共性,表明共发生(co-occurring)的NDDs具有潜在的遗传多效性(genetic pleiotropy)和共有病因。
细胞外基质,细胞基质局灶粘连,细胞骨架网络和Rho GTPase基因与CP高度相关(Extracellular matrix, cell–matrix focal adhesions, the cytoskeletal network and Rho GTPase genes are highly associated with CP)。我们鉴定了大量的、被预测存在有害变异的基因,并使用了一套工具来无偏差地发现与CP相关的生物通路和生物功能。我们基于STRING(宋红卫:STRING是一个常用的PPI数据库与分析工具)聚类(clustering)了439个假定的CP基因风险(补充数据集6-15),结果显示出比随机预测更强的连通性(greater connectivity than predicted by chance,enrichment=1.2, P=1.51×10-4),展示出一个包含有害变异的(基因)功能(互作)网络。我们又利用DAVID、MSigDB和PANTHER对其中的67,68个基因通过超几何分布检验进行了功能注释和通路表征。该方法展示了候选基因在分层的基因本体论(Gene Ontology,GO)和(生物)通路(KEGG/Reactome)的超几何统计检验,进而对功能和表达数据进行挑选和整理,以确定有意义的关联(curated functional and expression data to identify meaningful relationships)。与STRING的发现一致,该方法获得了所富集通路与功能中的多个基因集(false discovery rate (FDR)<0.05)(补充数据集6-15)。
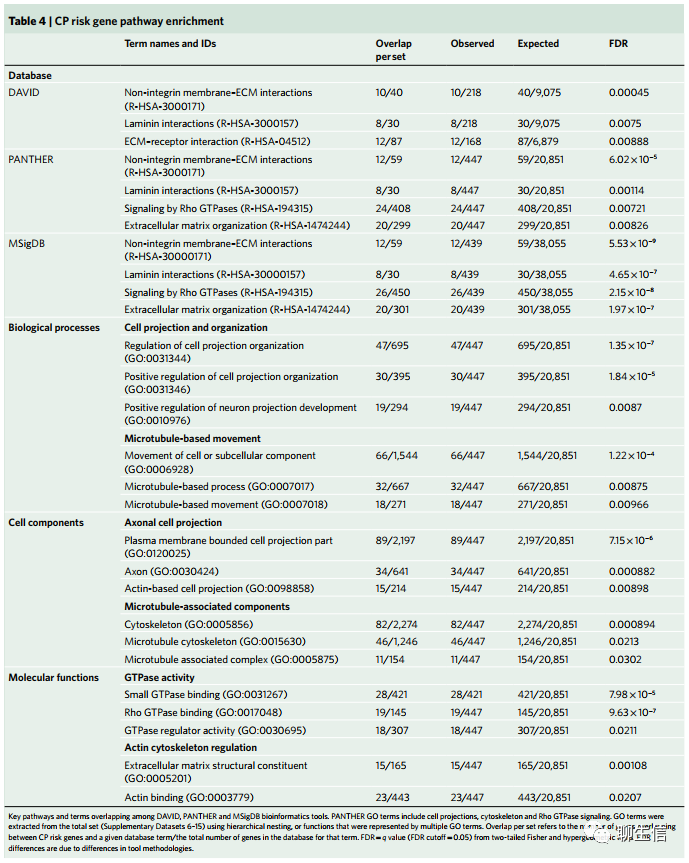
我们注意到由多种工具支持的相关功能的发现,包括所有三种算法确定的非整合素膜-细胞外基质(ECM)相互作用和层粘连蛋白相互作用途径。然后我们使用dcGO推断出GO Terms之间的层次关联(表4)。综合而言,这些发现表明了涉及:ECM(细胞外基质)生物学、细胞-基质相互作用(局部黏附)、细胞骨架动力学和Rho GTPase功能的富集。
来自Rho GTPase、细胞骨架和细胞投射通路的基因,可调控果蝇的神经运动发育(Genes from Rho GTPase, cytoskeleton and cell projection pathways govern neuromotor development in Drosophila)。随后,我们通过在果蝇中进行反向遗传筛选,独立评估了所富集通路在正常运动发育中的作用。类似的方法以前曾分别应用于果蝇和斑马鱼中对ASD和HSP的研究。我们的研究重点是存在害变异的基因,这些基因来自于我们的CP患者队列,包括GTPase、细胞骨架和细胞突/投射(cell projection)GO通路(GO Terms)。我们推测,我们的筛选可能首次揭示这些基因在神经运动发育中的关键作用。
我们选择了果蝇中具有保守性的同源基因(DIOPT≥5),且具有可用的分子特征的等位基因(完整结果和基因型见补充表9)。我们利用双等位基因状态下的亚型(或LoF)等位基因,帮助将表型映射到果蝇实验中感兴趣的基因。我们排除了可能导致混杂表型(如致死性)或具有先前描述的运动表型的基因,ATL1除外(作为阳性对照纳入)。优先考虑在大脑发育或NDD中已知有作用的基因。我们使用两个变异不符合有害性筛选标准的基因,作为阴性对照。总的来说,我们在幼虫中使用旋转试验,在成虫中使用负趋地性(或正趋光性)试验,筛选了22个与运动能力有关基因。
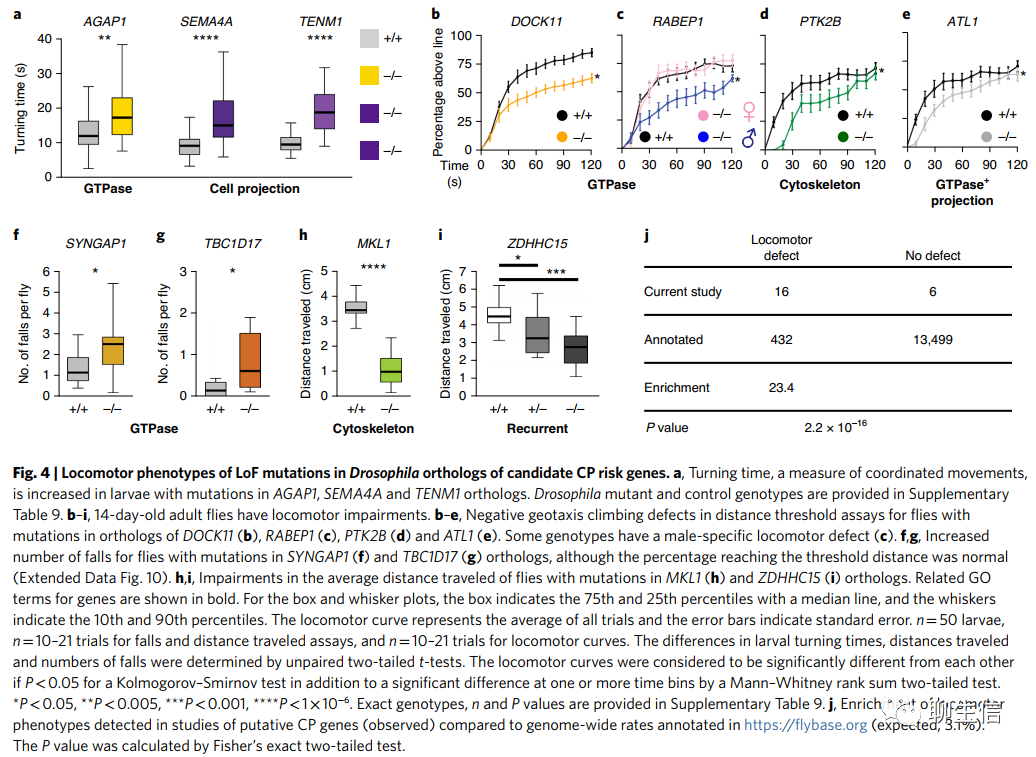
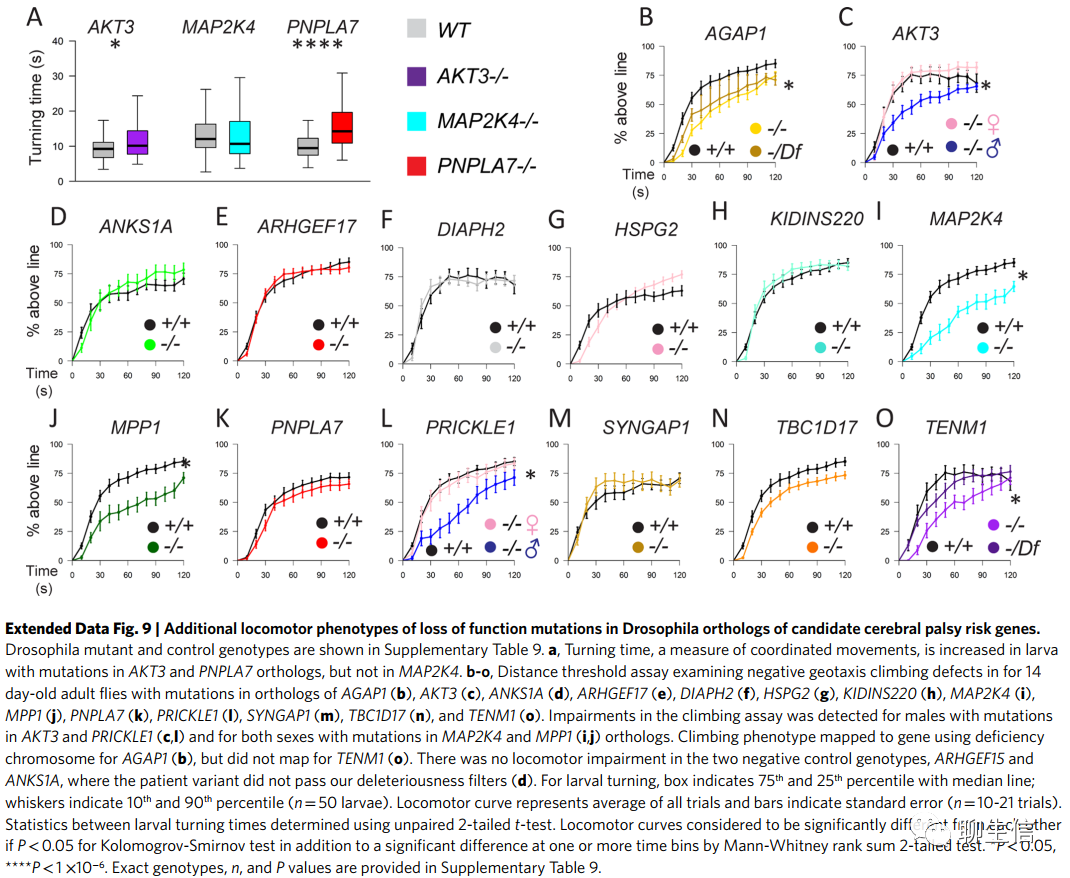
我们在GTPase信号转导调控基因(AGAP1, DOCK11, RABEP1, SYNGAP1和TBC1D17)、细胞骨架(MKL1和MPP1)和细胞投射(PTK2B, SEMA4A和TENM1)通路的突变体中发现了运动表型(图4)。 当对幼虫和成虫进行检测时,我们经常在两个时间点均发现运动表型,这表明缺陷出现在发育时期,并贯穿整个生命周期(补充表9)。有趣的是,我们发现了性别二态性的证据,AKT3、RABEP1或PRICKLE1/2同源基因突变的雄性果蝇表现出了运动缺陷,但雌性果蝇没有。
总之,我们发现71%(10/14)来自我们富集途径的基因在果蝇中表现出运动表型(图4和扩展数据图9)。相比之下,全基因组范围内,只有3.1%的注释果蝇基因已知会导致运动表型(enrichment=23.4, P=2.2×10-16; Fig. 4)。总之,我们的果蝇研究能够支持候选CP基因在运动发育的细胞骨架、Rho GTPase和细胞投射通路中的作用。
04 | 讨论
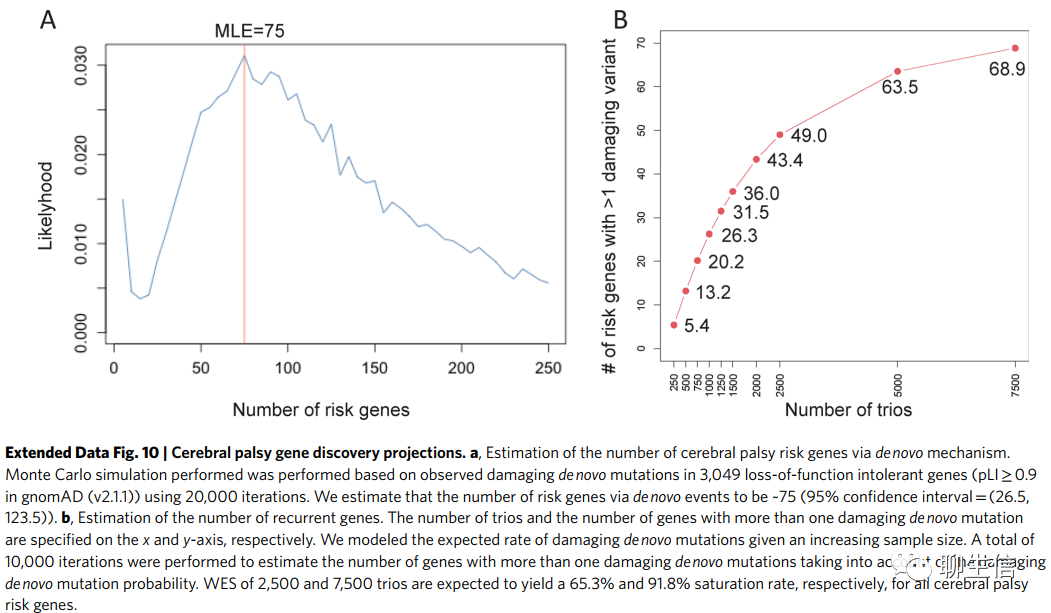
在过去,有害的基因组变异并未被认为是CP(脑瘫)的主要成因,但我们和其他人的发现挑战了这一信条。先前的研究表明,CNV和单核苷酸变异都与CP有关。在这里,我们对这些早期的发现进行了扩展,并在罕见的队列水平上提供了可靠的统计学证据,(发现)有害的单核苷酸变异是CP的一个独立风险因素。我们检测到的在队列范围内DNMs(新发突变)的富集,与“大多数CP病例是偶发的”这一观测结果一致(宋红卫:这意味着不能单靠某一个或某几个基因就能准确判断出CP的病因。这也是当下利用遗传变异去推测许多疾病成因的一大障碍。但并不意味着将来不会得到解决)。利用该队列中含有多重有害DNM的LoF(功能缺失)不耐受基因的分布(distribution of LoF-intolerant genes with multiple damaging DNMs),我们估计通过从头机制导致CP的基因数量为75个(95%置信区间为26.5–123.5;Extended Data Fig. 10a和Supplementary Note)。饱和度分析(Saturation analysis)估计,对于含有DNMs的CP风险基因,2500和7500个CP三人组的WES分别产生65.3%和91.8%的饱和(度),这表明随着更多的样本测序,会发现更多的CP(风险)基因(Extended Data Fig. 10b)(宋红卫:因为CP是散发的、偶发的,即:每个CP病人的致病位点和致病基因不尽相同)。因此,国际脑瘫基因组学联合会(ICPGC,https://www.icpgc.org)近期成立,旨在满足国际数据之间的共享和协作需要,以加快(CP风险基因)的发现速度。保守地说,我们估计我们队列中14%的病例可以通过有害基因组变异来解释(基于差异鉴定,DNMs为11.9%,RGs为2%)。相比之下,最近的估计表明,约6%的CP病例出现了急性分娩期/围产期缺氧缺血,这表明基因组突变是CP病因的一个重要的独立因素,而这在过去一直被忽视。
我们在队列中发现了已知疾病相关基因和以前与人类表型无关的基因的证据。RHOB和FBXO31中独立产生但相同的DNM的鉴定表明CP的单基因致病因素存在,但可能未被充分认识。我们对CP与其他NDD(神经发育障碍)的遗传相关性的平行检测,暗示了(它们之间具有)共同的易感(基因),如之前另一篇文献所述。在某些情况下,这可能反映了测量偏倚(ascertainment bias),因为在以前的其他NDD研究中,运动表型可能报道不足。在其他情况下,以FBXO31为代表,我们的发现可能代表了表型的扩展(phenotypic expansions)。最后,在某些情况下,NDD的临床表现可能被证明是多向性的(pleiotropic),早期神经发育的遗传损害(genetic disruption)表现出了多样性,这一点越来越为人们所知。至于其他NDDs,个别CP病例(的起因)可能证明是环境因素、遗传因素或两者的某种组合。然而,在NDDs中,环境对CP的贡献相对较好,CP可能是一种模型疾病(a model disorder),需在(个体的诞生、成长和)发育过程中研究基因与环境的相互作用。
运动回路连通性的改变被认为是CP病理生理学的一部分。通过整合正交证据线,包括复发性(recurrent)基因分析、体外和体内功能分析、队列范围内的网络生物学方法和果蝇运动研究,我们发现了支持ECM(细胞外基质)成分、细胞-基质局灶性粘附、细胞骨架组织和Rho GTPases在CP病因中作用的一致证据。已知这些过程驱动神经系统发育过程中细胞投射和延伸的保守过程(conserved process)。因此,根据已知的疾病和发育生物学,我们预测,参与神经发育模式形成的基因的破坏可能会改变CP的早期神经突生成和功能神经元网络的连接。需要进一步的研究来更具体地确定CP患者中发现的变异如何影响神经回路的发育。
我们的发现具有重要的临床意义。具体的遗传变异的发现可能为家系(分析)提供线索,并指导预防性保健和生育计划,如对复发风险进行(遗传)咨询(often quoted as ~1% for CP ,但遗传突变可能更高)。在某些情况下,在我们的队列中,对个体特定变异的鉴定导致了对治疗(方案)进行改变的建议,包括本来不会启动的个性化治疗,如:GNB86 (F068)的乙索妥昔胺(用药), CTNNB1(F066, GRA8913,F428)的左旋多巴(用药),和AMPD88 (F623)的5-氨基咪唑-4-羧基酰胺核苷(AICAr)(用药)(Supplementary Note)。
在不久的将来,我们的研究将能够克服样本量小的限制,进一步利用现有的临床数据来阐明基因型与表型的相关性(expand on genotype–phenotype correlations)。此外,随着更多关于CP遗传学致病因素信息的获得,会将可能的遗传原因分配给更多的个体病例。未来对特征明确的未选择的(well-characterized unselected)CP队列的研究将有助于确定遗传和环境因素对CP流行病学的真正影响。
总的来说,我们的数据表明,在评估个体CP病因时,基因组变异应与环境损伤一起考虑,这些考虑将具有重要的临床、研究和法医学(medico-legal)意义。 在不久的将来,基因组数据可能有助于对患者进行分层,并确定目前可用的医疗和(或)手术治疗的可能应答。 最后,随着时间的推移,通过CP的基因组研究确定的核心(生物)通路所产生的机制性理解(mechanistic insights),可能有助于指导几十年来没有新疗法出现的领域的治疗开发工作。
05 | 方法(原文截图)









关于人类遗传学知识、文献和数据分析技术,可关注和星标“聊生信”、“生信宝典”
往期精品(点击图片直达文字对应教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|




























后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集




















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









