






 研究人员利用ESM1b,一个6.5亿参数的蛋白质语言模型,预测人类基因组中错义变异的效应,超越现有方法。ESM1b在临床和实验数据集上表现出色,尤其在区分致病性和良性变异以及预测变异临床影响方面。
研究人员利用ESM1b,一个6.5亿参数的蛋白质语言模型,预测人类基因组中错义变异的效应,超越现有方法。ESM1b在临床和实验数据集上表现出色,尤其在区分致病性和良性变异以及预测变异临床影响方面。
编译 | 曾全晨
审稿 | 王建民
今天为大家介绍的是来自Chun Jimmie Ye和Vasilis Ntranos团队的一篇关于语言模型应用的论文。预测编码变异的效应是一个重大挑战。尽管最近的深度学习模型在变异效应预测准确性方面取得了改进,但由于依赖于近源同源物或软件限制,它们无法分析所有编码变异。在这里,作者开发了一个工作流程,使用ESM1b,一个拥有6.5亿参数的蛋白质语言模型,来预测人类基因组中约4.5亿个可能的错义变异效应。ESM1b在将约15万个ClinVar/HGMD错义变异分类为致病性或良性,并在28个深度突变扫描数据集中预测测量方面优于现有方法。

遗传变异的表型后果,即变异效应预测(Variant Effect Prediction,VEP),是人类遗传学中的一个关键挑战。改变蛋白质氨基酸序列的编码变异因其在疾病关联、机制理解和治疗可行性方面的丰富性而具有特殊的兴趣。大多数自然发生的编码变异是错义突变,将一个氨基酸替换为另一个。尽管在功能基因组学和遗传研究方面取得了进展,但区分蛋白质破坏性有害变异与中性变异仍然是一个挑战。此外,大多数人类基因存在可选择性剪接,同一变异可能对某些蛋白质异构体具有破坏作用,但对其他异构体中性,这取决于与蛋白质其余部分的相互作用。因此,大多数错义变异仍然是不确定意义变异(Variant of Uncertain Significance,VUS),限制了外显子组测序在临床诊断中的应用。对于影响多个氨基酸残基的编码变异,例如帧内插入缺失(in-frame indels),VEP甚至更具挑战性。
VEP的实验方法,如深度突变扫描(Deep Mutational Scans,DMS)和扰动测序(Perturb-seq),可以同时测量数千个变异体的分子和细胞表型。然而,这些内表型并不完全是相关临床表型的代理,而且在全基因组范围内难以扩展。相比之下,学习蛋白质的生物物理性质或进化约束的计算方法在理论上可以覆盖所有编码变异。尽管大多数计算方法是基于对致病性与良性变异的标记数据进行训练的,无监督的基于同源性的方法可以直接从多序列比对(MSA)中预测变异效应,而无需对标记数据进行训练。最近,一种名为EVE的无监督深度学习方法,实施了生成式变分自编码器,被证明在性能上优于监督方法。然而,由于它们依赖于MSA,基于同源性的方法仅提供一部分对齐良好的蛋白质和残基的预测。此外,由于同一基因的不同异构体具有相同的同源物,不清楚它们是否能够区分变异对不同异构体的影响。
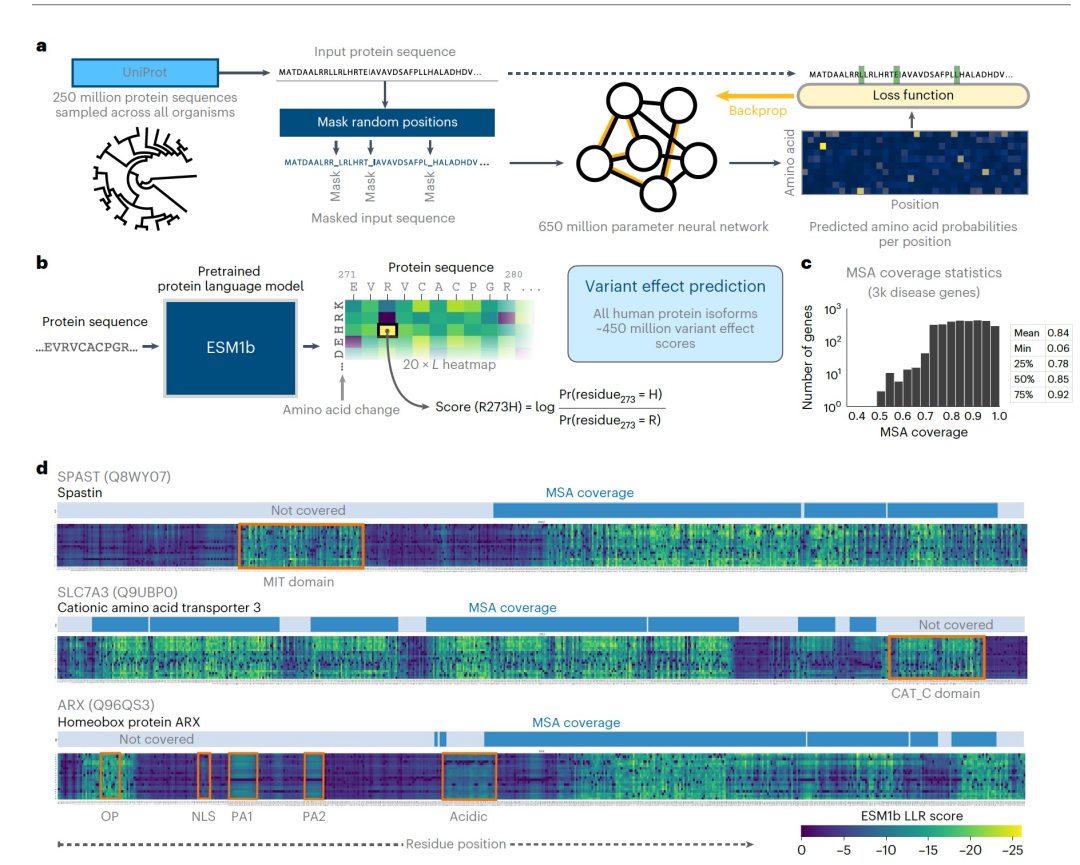
图 1
VEP的另一种深度学习方法使用蛋白质语言模型,这是一种源自自然语言处理的技术。这些是经过训练的深度神经网络,用于模拟通过大型蛋白质数据集(如UniProt)捕获的整个进化过程中已知蛋白质序列的空间(图1a)。值得注意的是,蛋白质语言模型不需要显式的同源性,可以估计任何可能的氨基酸序列的可能性。已经证明它们能够隐式地学习蛋白质序列如何决定蛋白质结构和功能的许多方面,包括二级结构、远程相互作用、翻译后修饰和结合位点。其中一个最大的蛋白质语言模型是ESM1b,它是一个公开可用的650百万参数模型,训练数据包括约2.5亿个蛋白质序列。已经证明,它能够在无需进一步训练的情况下预测与DMS实验结果相关的变异效应。
然而,ESM1b的使用受到了几个限制。首先,该模型的输入序列长度限制为1,022个氨基酸,排除了大约12%的人类蛋白质异构体。其次,虽然在32个基因(其中10个来自人类)的DMS数据上进行了评估,但目前尚不清楚该模型在全基因组范围内预测编码变异临床影响方面的表现如何。最后,使用ESM1b需要软件工程技能、深度学习专业知识和高内存GPU,这些因素共同构成了广泛使用的技术障碍。在这里,作者将ESM1b推广到任意长度的蛋白质序列,并用它来预测人类基因组中所有42,336个蛋白质异构体的约450 million个可能的错义变异效应。作者在三个不同的基准测试上评估,并将其与其他45种VEP方法进行了比较。
预测人类基因组中所有可能错义变异的效应
作者开发了一种改进的ESM1b工作流,并将其应用于获取所有42,336个已知人类蛋白质异构体上的约450 million个错义变异效应的完整目录。每个变异的效应分数是变异和野生型(WT)残基之间的对数似然比(LLR)(图1b)。与目前仅适用于一部分人类蛋白质和具有MSA覆盖率的残基的同源性模型(图1c)不同,ESM1b预测了每个可能的错义变异的效应。由ESM1b预测为有害的蛋白区域中的许多可能突变通常与已知的蛋白质结构域对齐(图1d)。如图所示,对于SPAST、SLC7A3和ARX,这些结构域可能位于MSA覆盖范围之外,不适合基于同源性的模型(图1d),但可能携带与疾病相关的变异。
ESM1b在临床和实验基准测试中表现优于其他VEP方法
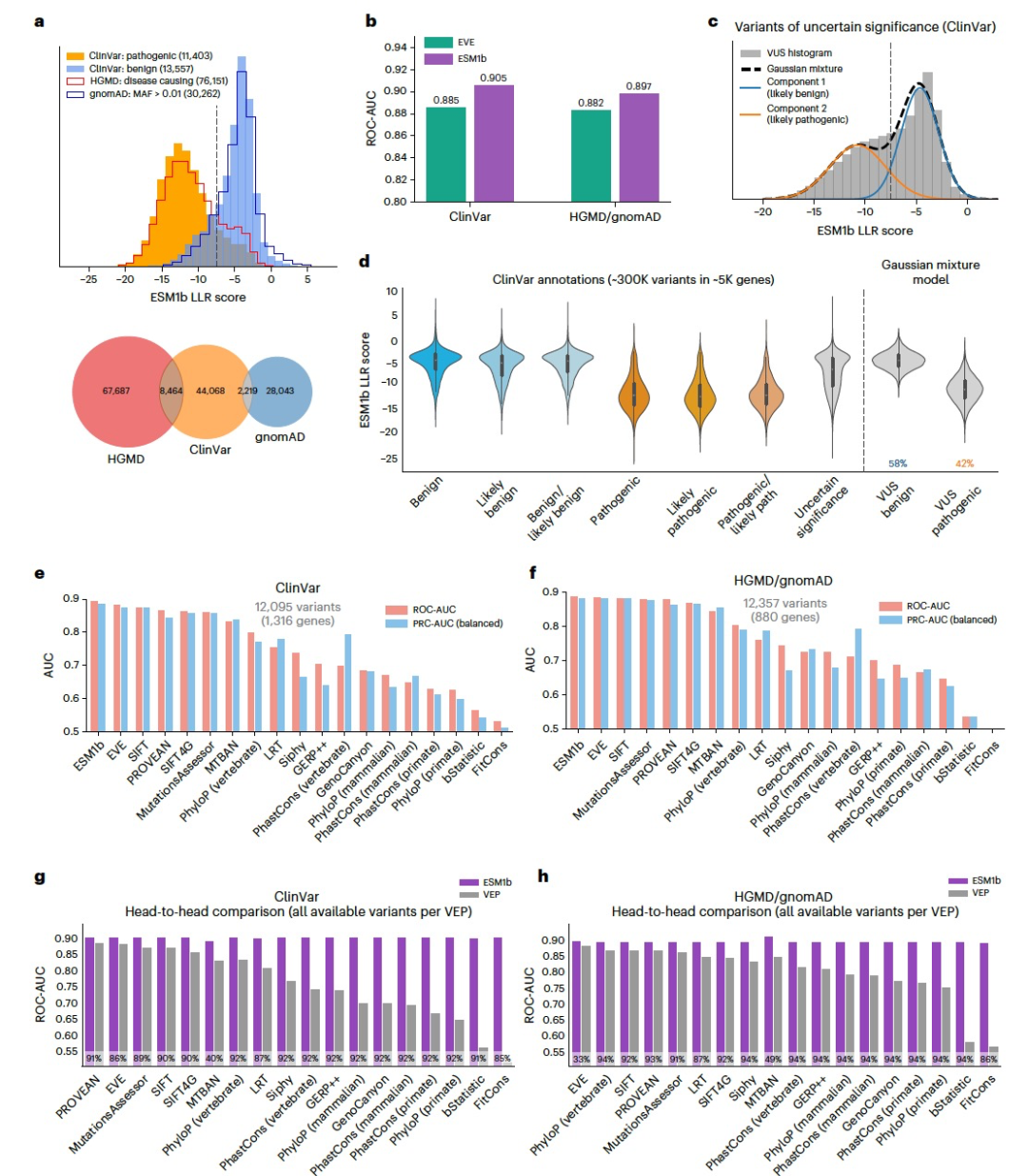
图 2
为了评估ESM1b在预测变异临床影响方面的性能,作者在两个数据集中比较了模型在致病性和良性变异之间的效应分数。第一个数据集包含了在ClinVar中注释的致病性和良性变异,第二个数据集包括了HGMD中被注释为致病性的变异,以及gnomAD中的良性变异(定义为等位基因频率大于1%)。ESM1b效应分数的分布在这两个数据集中显示出致病性和良性变异之间的显著差异(图2a)。此外,在这两个数据集中,致病性和良性变异显示出一致的分布,表明预测结果具有很好的校准性。使用LLR阈值为-7.5来区分致病性和良性变异,在这两个数据集中的真阳性率分别为81%和82%。将ESM1b与EVE作为变异致病性的分类器进行比较,ESM1b在区分ClinVar中的19,925个致病性和16,612个良性变异(跨越2,765个基因)方面获得了0.905的ROC-AUC分数,而EVE的分数为0.885。在HGMD/gnomAD中(涵盖1,991个基因,包括27,754个致病性和2,743个常见变异),ESM1b获得了0.897的ROC-AUC分数,而EVE的分数为0.882(图2b)。
在确认ESM1b作为变异致病性分类器的高准确性后,作者尝试预测ClinVar中VUS的效应。为此将ESM1b效应分数在VUS上建模为具有两个成分的高斯混合分布(图2c)。这两个拟合的分布与注释的致病性和良性变异的分布吻合良好(图2d)。根据该模型,作者估计ClinVar中约58%的错义VUS是良性的,约42%是致病的。除EVE之外,作者还将ESM1b与其他44种VEP方法进行了比较,包括来自Database for Nonsynonymous SNPs’ Functional Predictions(dbNSFP)的所有功能预测方法和保守性评分。在临床基准测试比较中,作者只考虑了那些(1)未在临床数据库(如ClinVar和HGMD)上进行训练,或未使用来自这些训练过的方法的特征,并且(2)不使用等位基因频率作为特征的方法,因为等位基因频率通常用于将变异标记为良性。在这46种方法中,有19种(包括ESM1b和EVE)满足这些无偏比较的标准。在所有19种方法报告的变异集合中,ESM1b在ClinVar和HGMD/gnomAD上均优于其他所有方法(图2e、f)。类似地,ESM1b在其各自报告的变异集合上也分别优于每种单独的方法(图2g、h)。所有两两比较的结果在统计学上都具有显著性,P值小于0.001。
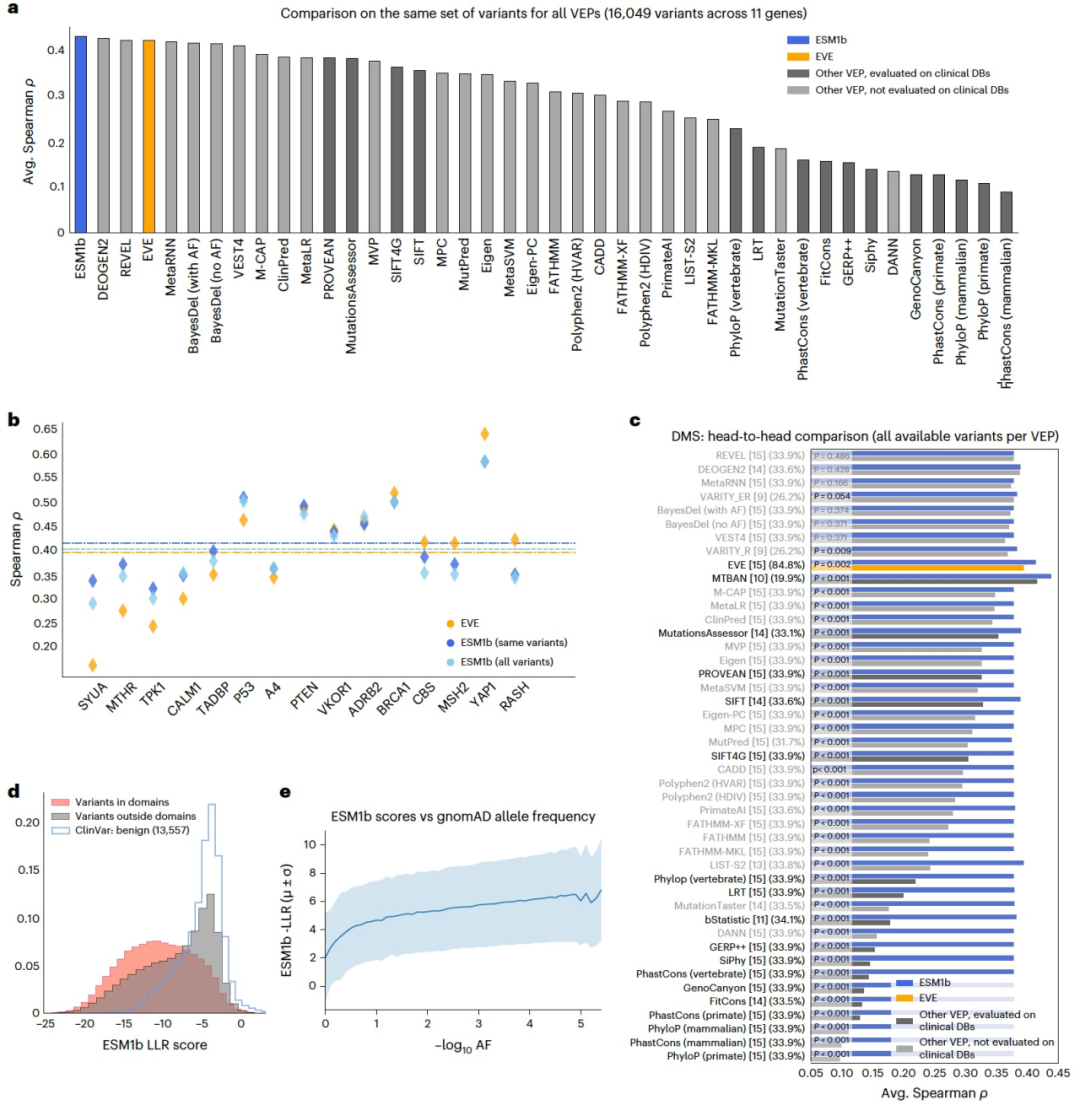
图 3
作者进一步比较了这46种VEP方法在预测DMS实验测量结果方面的能力。完整的DMS基准测试包括28个实验,涵盖了15个人类基因(在76,133个变异上的166,132个实验测量)。作者将43种方法与由这些方法报告的11个基因中的16,049个变异的子集进行了比较。ESM1b以平均斯皮尔曼相关系数0.426的得分位居首位,其效应分数与实验测量之间的关系图如图3a所示,其次是DEOGEN2(0.423)、REVEL(0.419)和EVE(0.418)。DEOGEN2和REVEL是监督方法,而EVE,就像ESM1b一样,是一种无监督方法。将ESM1b与EVE直接与具有EVE分数的64,580个变异(跨足15个基因)进行比较,结果呈现出类似的趋势(图3b)。同样,ESM1b在每种方法报告的变异集合上都优于其他45种方法(图3c),其中有37种方法的比较在统计学上显著。另外两项附加分析进一步证明了ESM1b预测的功能解释。首先,如个别示例所示(图1d),位于结构域内的错义变异具有更负面(有害)的效应分数,而位于结构域外的变异类似于良性变异(图3d)。其次,ESM1b效应分数与等位基因频率很好地吻合,常见变异被预测为较不具有破坏性(图3e),这与常见变异通常被认为是良性的一致。
ESM1b可以预测变异对蛋白质异构体的影响
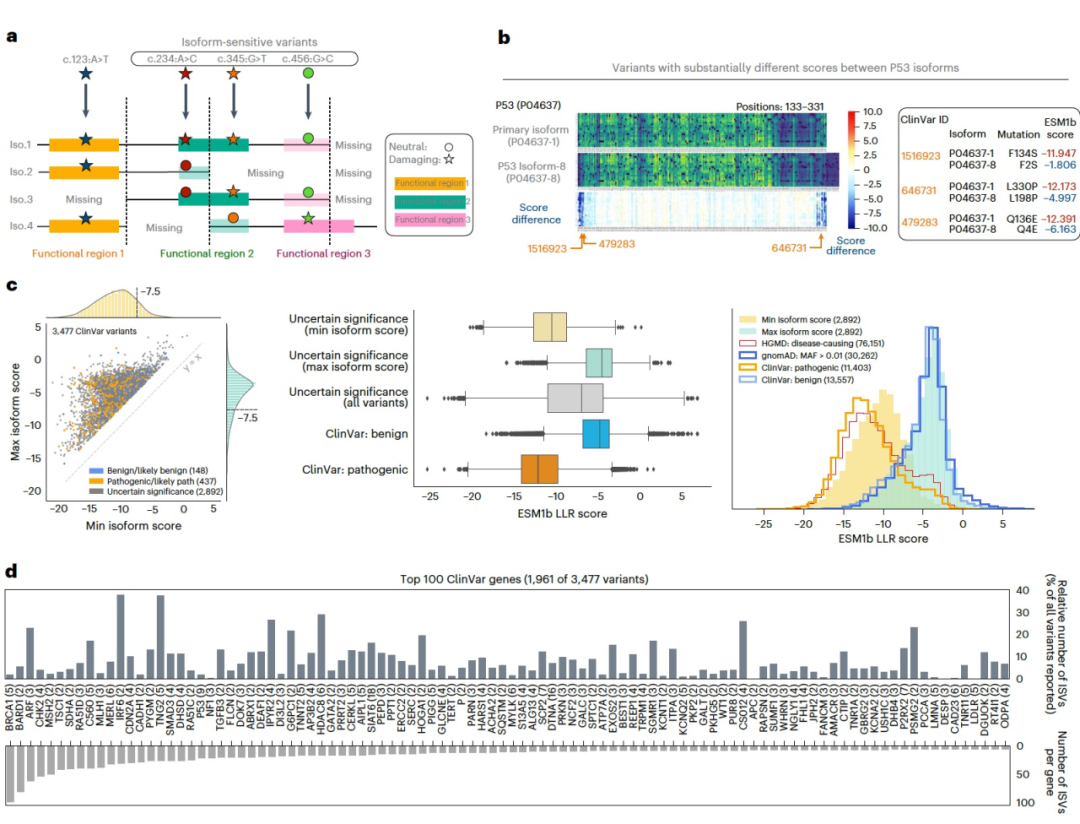
图 4
作为一个蛋白质语言模型,ESM1b在输入的氨基酸序列的上下文中评估每个变异,允许在不同蛋白质异构体的上下文中评估相同的变异。一个变异可能对某些异构体具有破坏性,但对其他异构体没有,这可能是由于与可选择性剪接结构域的相互作用(图4a)。例如,比较P53的主要异构体和一个较短的异构体之间的ESM1b分数,作者发现170个变异(主要位于剪接交界处附近)的分数差异很大(LLR差异>4),其中包括三个ClinVar变异,被注释为VUS(图4b)。在ClinVar中发现了3,477个错义变异,其在异构体间预测的效应有显著差异(LLR标准差>2)(图4c)。值得注意的是,作者只考虑了经过审查和手动筛选的蛋白质异构体。这3,477个变异中,包括148个(4%)良性或可能良性,437个(13%)致病性或可能致病性,以及2,892个(83%)VUS。有趣的是,在考虑最具有破坏性的异构体时,这些VUS的效应分数分布与致病性变异的分布相似;而在考虑最不具有破坏性的异构体时,这些VUS的分布与良性变异相似(图4c)。与P53类似,许多临床重要的基因在不同异构体间具有高效应分数方差的ClinVar变异,包括BRCA1、IRF6和TGFB3(图4d)。
结论
全面的评估显示,ESM1b在区分ClinVar和HGMD/gnomAD中的致病性和良性变异,以及预测DMS实验报告的效果方面,表现优于其他最先进的VEP方法。作为一种不明确依赖同源性的蛋白质语言模型,ESM1b在VEP方面提供了几个额外的优势。作为一种无监督方法,ESM1b在临床或人群遗传学数据集中,不存在从训练集到测试集的信息泄露风险,从而可以进行准确和无偏的评估。与基于同源性的方法相比,ESM1b的预测更简单且更快速,因为一旦训练了通用模型,只需要一个输入序列。值得注意的是,蛋白质语言模型可以为每个可能的氨基酸序列提供预测,并适用于所有编码变异。在研究中,已经证明了ESM1b的普适性,包括(1)MSA覆盖范围之外的变异,(2)对不同蛋白质异构体的不同影响的变异,(3)帧内插入缺失和(4)终止密码子变异。
参考资料
Brandes, N., Goldman, G., Wang, C.H. et al. Genome-wide prediction of disease variant effects with a deep protein language model. Nat Genet (2023).
https://doi.org/10.1038/s41588-023-01465-0
往期精品(点击图片直达文字对应教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
















































 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









