






点击蓝字 关注我们
必需基因特性表征与相关数据库开发的研究进展

iMeta主页:http://www.imeta.science
综 述
● 原文链接DOI: https://doi.org/10.1002/imt2.157
● 2024年1月2日,天津大学高峰团队在iMeta在线联合发表了题为 “Recent advances in characterization of essential genes and development of a database of essential genes” 的研究文章。
● 本文总结了必需基因的相关研究进展,介绍了DEG数据库及主要应用,并基于最新版本DEG 15.0进行了统计分析。应当注意的是必需基因是一个动态概念,而不是一个二元概念,这为必需基因未来的研究发展带来了机遇和挑战。
● 第一作者:梁雅婷
● 通讯作者:高峰(fgao@tju.edu.cn)
● 主要单位:天津大学理学院物理系、天津大学合成生物学前沿科学中心和系统生物工程教育部重点实验室、天津化学化工协同创新中心
亮 点
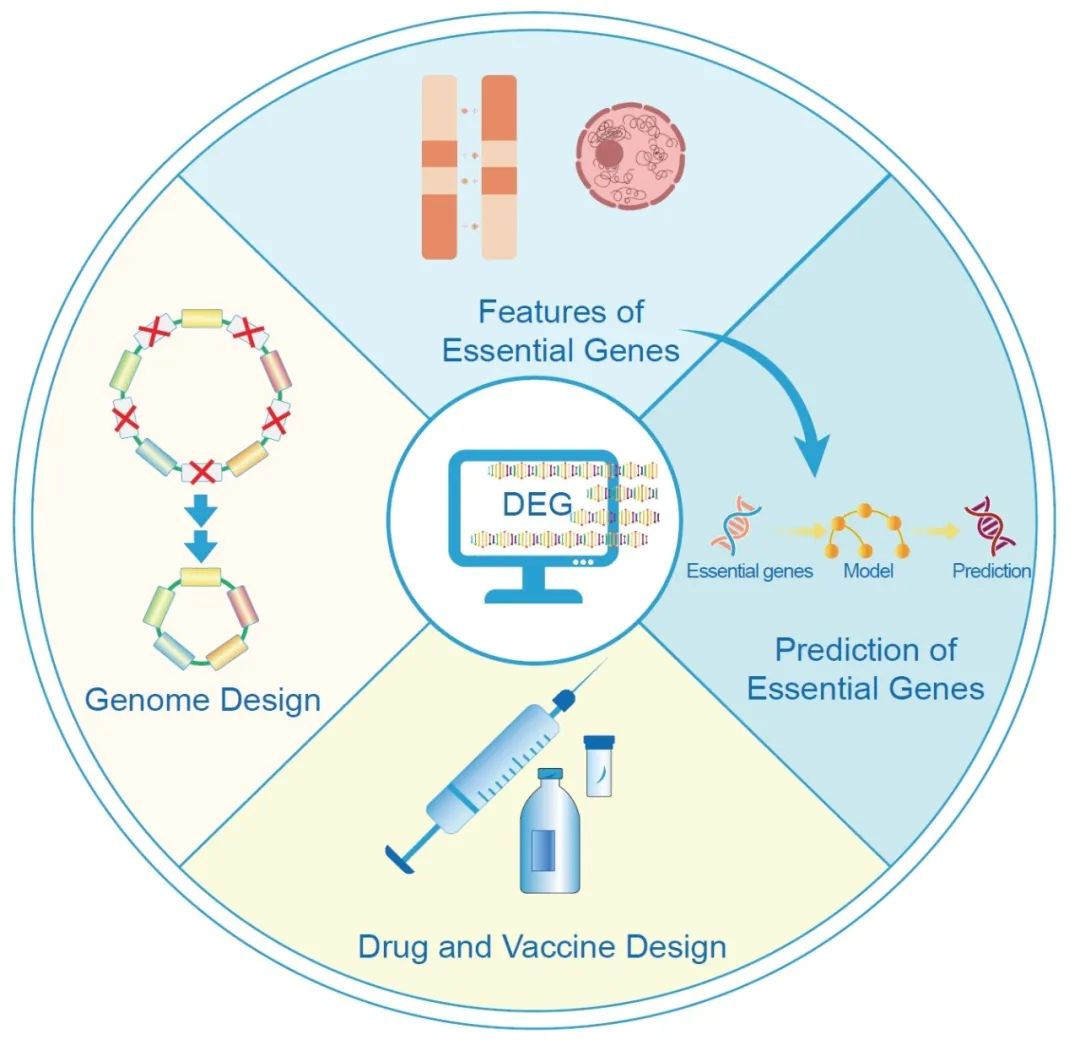
● 必需基因对生物体的生存和发育至关重要,目前已通过多种实验手段鉴定出了大量生物体的必需基因,为研究它们的特性和开发预测算法提供了宝贵的资源;
● DEG数据库收集了大量必需基因的实验结果,是开展必需基因相关研究的可靠资源,方便研究人员快速获取特定物种的必需基因信息,并在必需基因特征分析和预测、药物和疫苗开发,以及人工基因组的设计和构建中进行应用;
● 必需基因的定义依赖于研究背景,而非简单的二元分类。通过整合不同实验结果,对必需基因进行定义和分类具有挑战性,但也为探索基因相互作用机制等相关研究提供了可能性。
摘 要
在过去的几十年里,对必需基因的研究引起了人们广泛的兴趣。这些基因对于生物体在特定环境条件下的生存至为关键,在合成生物学和医学领域具有重要的应用价值。随着技术的不断发展,已通过实验手段获得了越来越多关于必需基因的数据。与此同时,各种与必需基因相关的计算预测方法、数据库和网络服务器相继出现。为了促进必需基因的研究,我们实验室建立了一个必需基因数据库——DEG,为必需基因特征分析和预测、药物和疫苗开发,以及人工基因组的设计和构建提供了重要参考。在本文中,我们总结了必需基因的相关研究进展,还介绍了DEG数据库及主要应用,并基于最新版本DEG 15.0进行了统计分析。应当注意的是必需基因是一个动态概念,而不是一个二元概念,这为必需基因未来的研究发展带来了机遇和挑战。
视频解读
Bilibili:https://www.bilibili.com/video/BV1X5411i7Pt/
Youtube:https://youtu.be/j22ihy62Wb8
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
引 言
必需基因是生物体在特定环境条件下生存所必需的基因,是分子生物学和遗传学的重要概念之一。基因的必需性在相关的理论和应用研究中都十分关键。对必需基因的鉴定十分重要,因为这些信息对生命科学、药理学和合成生物学等领域具有实际应用价值。然而,基因的必需性受多种因素影响,包括生长条件、发育阶段和遗传背景等。在不同的实验条件下,同一生物体可能展现出不同的基因必需性,且这些必需性可能随着进化而发生变化。基因的必需性是一个动态的、需要持续评估的量化特征,特定条件下的必需基因的作用不应被忽视。
通常,必需基因的识别依赖于实验手段。1999年,通过在生殖道支原体上进行全域转座子突变实验,首次确定了生物体在基因组水平上的一组必需基因。随后,科研人员对各种生物体中必需基因展开了广泛研究。识别必需基因实验技术的发展促进了必需基因数据的积累。识别必需基因最常用的实验方法包括单基因敲除、RNA干扰、反义RNA、转座子突变和CRISPR/Cas9等。除了实验方法,必需基因的计算预测方法也相继出现,进一步促进了必需基因的识别。最常用的计算方法包括基于比较基因组学、基于约束的预测方法和基于机器学习的预测方法,它们为未来的必需基因研究提供了重要参考。
近年来,必需基因的研究已在各个领域进行。例如,了解必需基因的功能对于发现最小基因组的组成至关重要,这是合成生物学中重要的研究内容之一。研究必需基因可以加速构建具有特定表型特征的生物,并促进药物和疫苗的开发。此外,它们在细菌生命中不可或缺的作用使病原体必需基因编码的蛋白可以作为潜在的药物靶标。因此,在多数细菌中保守的某些必需蛋白被视为有前景的广谱药物靶标,若仅在某种细菌中是必需蛋白,则为特定物种特异性靶标的可行候选。此外,还可以通过现有必需基因数据构建模型,来预测潜在基因的必需性。
随着必需基因数据量的不断增加,基于实验或预测得到的必需基因数据的数据库和在线服务应运而生,为必需基因相关研究提供了便捷可靠的参考。必需基因数据库(DEG)于2004年建立,并随着实验数据的积累不断更新。该数据库囊括了从多种实验方法获得的全基因组规模的必需基因数据。到目前为止,DEG数据库已成为必需基因相关研究的重要参考,根据Web of Science数据,该数据库已累计被引用超过1100余次。
在本综述中,我们简要概述了必需基因相关的技术发展、数据库等网络服务的构建、必需基因的应用,并强调了基因必需性是特定于环境的概念。此外,我们还介绍了DEG数据库在实际研究中的重要应用,并提出了未来DEG数据库的发展方向。
必需基因的定义
必需基因对生物体的生存至关重要,因此被视为生命的基础。然而,不同物种和研究中必需基因的估计比例存在显著差异。同一物种内的不同研究显示,某些基因在一个菌株中可能是必需的,而在另一个菌株中则不是;或者某些基因在一种生长条件下是必需的,而在其他条件下则不是,这表明基因的必需性不是其固有属性。造成这种变化的原因多种多样,包括实验方法、条件的不同,甚至实验误差的影响。因此,“必需基因”这一术语高度依赖于研究背景。只有当生物体存活的环境被精确定义时,一个基因才能被归类是否为必需。细胞对特定基因或基因产物的依赖受其外部环境和遗传背景的影响。因此,基因的必需性可能因这些因素而变化,并随着每次删除而变化。这些基因被称为“条件性”的必需基因。例如,一些基因被识别为“保护性必需”,因为它们在移除另一个基因后可能变得非必需。这通常是因为前者编码了对后者基因毒性效应的保护功能。相反,第二个基因的丧失可能使一个非必需基因变得必需(合成致死)。合成致死最初是在研究果蝇和酵母时发现的,其中单独失活两个基因中的任何一个对细胞存活的影响都很小。然而,同时干扰两个基因或多个独立基因的表达,包括突变、过表达和基因抑制,可以导致细胞死亡。此外,基因产物可以形成复合物,其中非必需基因有助于必需功能。例如,在涉及繁殖的出芽酵母的蛋白质编码基因中,已识别出五组潜在编码具有必需功能蛋白质的非必需基因。营养条件也可以影响必需基因,因为携带失活非必需基因的突变菌株在最佳条件下可能对细胞表型产生最小或可忽略的影响,在次优条件下可能导致严重损害或生存能力丧失。然而,有证据表明在富营养培养基中生长的许多非必需基因对适应替代生长条件很重要。此外,不同的实验方法可能产生不同的结果。例如,与基于RNA干扰(RNAi)方法相比,基于CRISPR方法在人类细胞系中识别出的必需基因更多。考虑到这些因素,基因的必需性可能是一个定量特征,而不是简单的必需/非必需二分类,需要更加标准化的定量方法来衡量。研究表明,“条件性”的必需基因通常表现出类似于非必需基因的特征。这归因于与其他必需基因相比,此类基因通常旁系同源基因数量较多,共表达水平较低,几乎不编码蛋白质复合物成分。这一结果解释了“条件性”的必需基因和“永久性”必需基因之间的区别。基于这些特征,研究者已开发出一种随机森林预测模型,用于识别有条件的必需基因。
最近的研究发现,细菌和酵母可以通过基因组变化和适应性突变来适应环境,这些突变可以在条件性必需基因失活的情况下恢复细胞功能。这些研究表明,尽管失去了必需基因的功能,基因的必需性取决于细胞获得突变和恢复增殖的能力。这表明必需性不再仅仅是基因属性,而是细胞属性,将必需性归因于细胞途径而非基因本身。这些发现为定义和研究必需基因提出了新的研究方向和挑战。
必需基因的鉴定
目前,确定必需基因主要有两种方法:实验方法和计算方法。实验方法可以在不同实验条件下为必需基因提供具体的结果(图1)。然而,这些方法可能成本高昂且劳动强度大。因此,利用计算机预测必需基因近年来得到了较快发展。
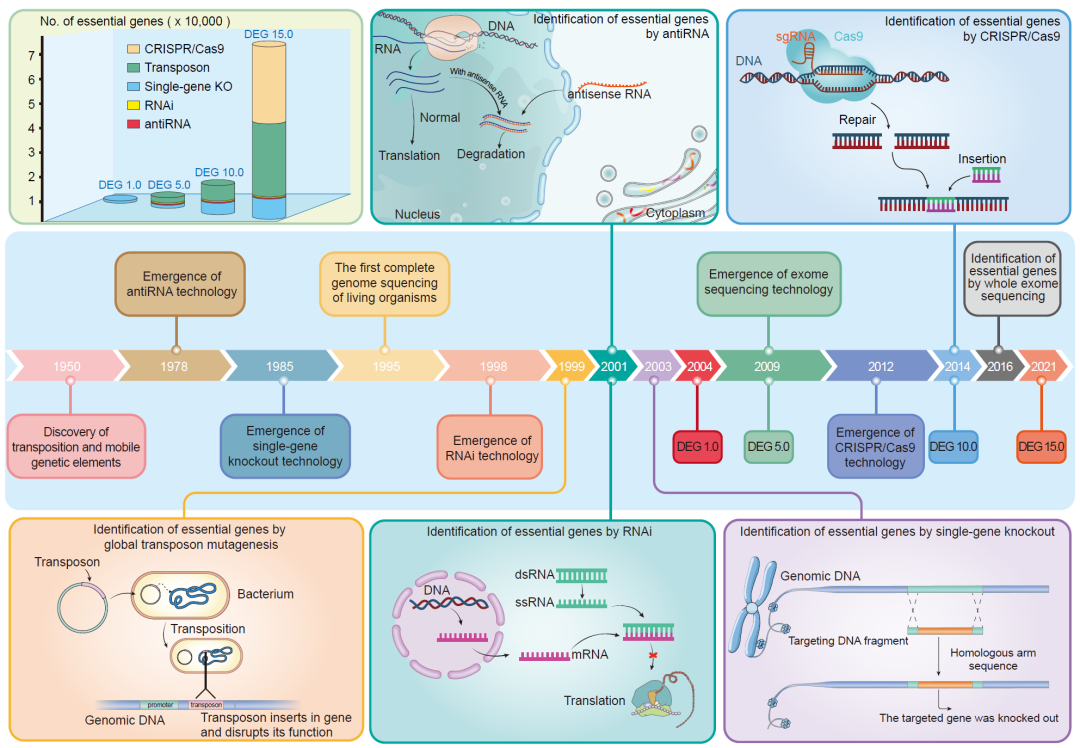
图 1. 必需基因相关技术发展时间轴
此图展示了一些重要的技术发展及其在必需基因研究中的最早应用。还显示了DEG数据库不同版本的更新时间,以及每个版本通过不同方法获得的数据量。
基于实验的方法
在分子生物学领域,鉴定必需基因有着悠久的历史。1951年,Horowitz和Leupold提出蛋白质的一些主要成分可能对生命至关重要。在“基因组时代”之前,早期的突变诱导技术涉及使用化学或物理试剂在生物体的基因组中诱导随机突变。通过诱导随机化学突变并分析后代存活率来预测必需基因。例如,研究表明,大约50%、15%和12%的果蝇、秀丽线虫和酿酒酵母的基因组是必需的。尽管如此,当时的实验技术不足以鉴定具体的必需基因。在20世纪末,发展了转座子技术、单基因敲除、反义RNA和RNA干扰等技术,为基因层面的研究提供了更多可能性。1995年,首次获得了流感嗜血杆菌和生殖道支原体的完整基因组序列。获得一个生物体的完整基因组序列是获取完整基因列表的先决条件。在接下来的两年中,大肠杆菌、枯草杆菌和酿酒酵母等模型生物的完整基因组也相继公布。1999年进行了第一个鉴定生物体必需基因集的实验,该实验通过全基因组转座子突变验证了生殖道支原体生存所需的最小基因集。转座子插入检测必需基因的原理基于转座子的随机插入及其对基因中断的影响。通过分析转座子中断基因的插入位点和表达,可以确定必需基因及其对生长的影响。随后,开发了通过针对单基因敲除、RNA干扰和反义RNA来鉴定必需基因的方法,使研究人员能够探索各种生物体中基因的必需性。单基因敲除实验涉及移除一个基因以观察表型变化。对于全基因组研究,这个过程必需重复多次,这需要全面的基因组注释。RNA干扰技术识别必需基因的原理是,通过引入与目标基因信使RNA(mRNA)互补的小干扰RNA(siRNA),形成双链结构,进而引发目标mRNA的降解,从而抑制相应蛋白的表达。反义RNA技术识别必需基因的原理是,通过引入与目标基因mRNA互补的单链反义RNA分子,这样做可以与目标mRNA形成双链结构,从而阻碍其表达,实现基因表达的抑制。
在必需基因研究的早期阶段,主要关注点是微生物。动物基因干扰的努力因动物组织培养中同源重组的固有低效而受阻。第一个突破是发现同源重组在来自小鼠囊胚的胚胎干细胞中更为有效,促进了1989年第一只基因敲除小鼠的研究。2003年,研究者对秀丽线虫进行了首次全基因组RNAi筛选,系统性地定义了突变体表型。随后,这项技术迅速应用于哺乳动物细胞,几个小组已经生成了涵盖人类和小鼠基因组的RNAi库,用于基因必需性筛选。RNAi已成为哺乳动物必需基因研究的主要方法。然而,不能忽略其非靶向效应和不完全的基因功能丧失限制。
下一代测序技术的出现使得快速获取各种物种的全基因组序列成为可能。方便的测序数据访问促进了引入处理遗传变异的其他方法。转座子测序(Tn-seq)是一种新兴技术,结合了转座子突变和高通量测序,并使用在目标生物体中构建的高密度转座子插入库,通过高通量测序对其整个基因组进行功能分析。如TraDIS、INSeq、HITS和Tn-seq等技术已广泛用于微生物中必需基因的检测。Tn-seq的应用通过纳入必需基因组元件(包括非编码RNA),而不仅仅关注蛋白编码基因,加深了对必需基因的理解。此外,Tn-seq可用于在体内、体外的各种实验条件下筛选必需基因,而不局限于培养条件。Tn-seq显著增加了在不同条件下鉴定的必需基因数量。
全基因组必需性筛选阐明了几个生物过程的分子基础。然而,基于RNAi的筛选常常受到非靶向效应和基因敲低而非完全丧失功能的困扰,这限制了我们对人类细胞中必需基因的了解。然而,CRISPR/Cas9系统的出现彻底革新了哺乳动物细胞基因组编辑。从根本上说,这个可编程的DNA内切酶由两部分组成:源自链球菌的Cas9蛋白(或其他物种的类似蛋白),以及引导内切酶活性至目标DNA序列的单一导向RNA。CRISPR诱导序列特异性DNA双链断裂,导致移码插入/缺失,从而导致蛋白功能的完全丧失。这项技术使得在酵母、植物和动物中进行经济高效的基因编辑成为可能,对人类细胞编辑产生了重大影响。此外,Cas9的催化失活版本(dCas9)可以用于通过单一导向RNA定位特定DNA序列,这被称为CRISPR干扰(CRISPRi),或用于激活基因表达,称为CRISPR激活(CRISPRa)。Cas9和dCas9已被用于绘制人类细胞系中必需成分的组成。2015年,三篇论文同时报道了不同人类细胞类型中必需基因的全基因组鉴定。此外,全外显子测序是另一项突破,它使用序列捕获或靶向扩增技术捕获和富集整个基因组外显子区域的DNA。与全基因组重测序相比,全外显子测序主要针对外显子区域的基因序列,具有更深入的覆盖和更高的数据准确性,以快速鉴定体内的人类必需基因。
基于计算的方法
考虑到实验方法的复杂性、高成本、劳动力和时间成本,计算方法通常被作为实验方法的补充,以最小化必需性分析所需的资源。通过实验鉴定的必需基因数量的增加为计算手段提供了参考。通常,有三种方法被用来确定基因的必需性:基于比较基因组学的、基于约束的和基于机器学习的方法。
最初,必需基因预测是通过依赖同源性的比较基因组学方法进行的。同源性映射发生在一个生物体内被复制的基因(旁系同源基因)或多个不同生物体中相关基因(直系同源基因)之间,这些基因是物种分化的产物。同源性映射通过比较多个生物体的序列,并根据一定的阈值确定它们的序列相似性。这种方法已被用于预测诸如支原体、棒状杆菌、疟原虫和布鲁氏菌等物种的核心基因。一个主要挑战是进化距离对比较基因组分析结果的显著影响。尽管必需基因在细菌中通常具有高度的进化保守性,但跨物种的保守基因并不总是必需基因,这使得基于同源性的方法在预测必需性方面效果较差。以前的大规模分析显示,只有少数基因在生命树上是保守的,这意味着许多必需基因是物种特异性的。
基于约束的预测方法利用基因组规模的代谢网络来阐明生物体内代谢途径的生物学。这种方法依赖于基于基因组测序和注释重建的代谢网络的约束建模技术,以研究网络的结构、功能和相互作用。通量平衡分析(FBA)是最广泛使用的基于约束的方法,用于分析代谢网络特性。它通过将质量平衡约束应用于化学计量模型,预测稳态条件下代谢物的流量。使用FBA预测必需基因涉及模拟基因敲除并评估其对网络的影响。基于约束的模型已在三域系统的生物体上构建,并促进了基因必需性的研究。FBA的计算成本较低,因为它不需要动力学参数。然而,FBA也有显著的局限性。首先,它只能预测代谢基因的重要性。此外,与能够结合稳态和动态分析的能力不同,FBA需要酶动力学数据来评估瞬态条件下基因组规模代谢反应的活性。最后,FBA通常需要酶反应来解决代谢模型中的局限性,有时可能与实验数据不一致。这取决于经验建模,在某些情况下,参数预测可能具有挑战性。
目前,最广泛使用的必需基因预测方法基于机器学习算法的构建,使用从必需基因的实验结果分析中得出的特征。通常,必需基因的特征可以分为两类:序列相关和背景相关(表1)。
表1. 必需基因的特征汇总
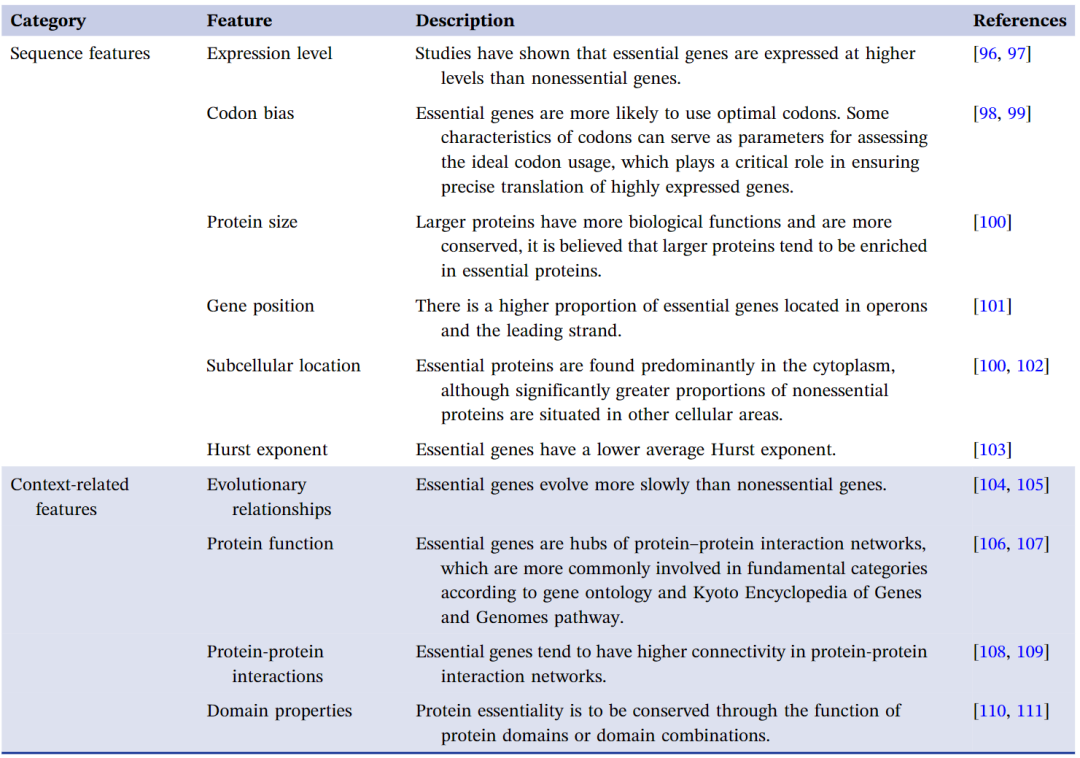
对必需基因特性的研究为使用机器学习预测必需基因提供了参考。通常,机器学习中预测模型的开发涉及以下步骤:构建训练和测试数据集(必需/非必需基因数据)、特征选择(必需基因的不同特征)、选择和设计机器学习算法,以及评估模型预测性能。各种研究已经使用基因组和蛋白质特征来开发和训练用于预测基因必需性的分类器。多年来,已成功开发了基于基因组特征的多种算法模型,用于理论鉴定必需基因。例如,范等人开发了一个结合必需基因蛋白质-蛋白质相互作用(PPI)和亚细胞定位的SCP算法。这种方法结合了基于基因表达数据的改进PageRank算法、加权亚细胞定位和加权亚细胞定位的皮尔逊相关系数。此外,还出现了预测非编码区域必需基因的模型。张等人使用基于metapath-guided的随机游走开发了iEssLnc模型,这是第一个估算长非编码RNA(lncRNA)基因必需性的模型。可以推断出,准确预测需要“好”的数据和高效的机器学习技术。监督学习、半监督学习、无监督学习和强化学习是常用的机器学习技术。然而,基因必需性预测通常被建模为在监督学习下的分类问题。
深度学习是人工智能中机器学习的一个子集,其中神经网络可以以无监督的方式从非结构化或未标记的数据中学习。最近,深度学习已被用于预测必需基因。例如,Deeply Essential是一个深度神经网络,仅使用序列信息来预测必需基因。与之前使用聚类和欠采样数据集的方法相比,这个模型实现了更高的灵敏度和准确性。另一种必需性预测的深度学习模型采用了不同的方法,使用一个自动学习生物特征的框架,而不需要先验信息。这个网络利用基因表达、亚细胞定位和PPI网络的信息来学习拓扑特征。然而,将深度学习应用于基因必需性预测有两个主要缺点:(i) 深度神经网络需要大量数据进行训练,以优于传统的机器学习算法;(ii) 深度学习模型中调整超参数的过程复杂。
虽然应用机器学习方法方便,但它也面临着一些挑战,如预测质量难以衡量和无法在特定实验背景下泛化。此外,考虑到必需基因的定义是特定于研究背景的,因此在定义训练ML模型的标准时应谨慎,这取决于研究的目的。此外,特征的选择和组合可能会影响预测性能,且没有确定的方法来为不同的生物体选择适当的特征。最后,对于研究不足的物种,选择研究物种内数据受限于已知必需基因的数量较少,而使用跨物种数据可能导致准确性降低。
相关数据库等网络服务
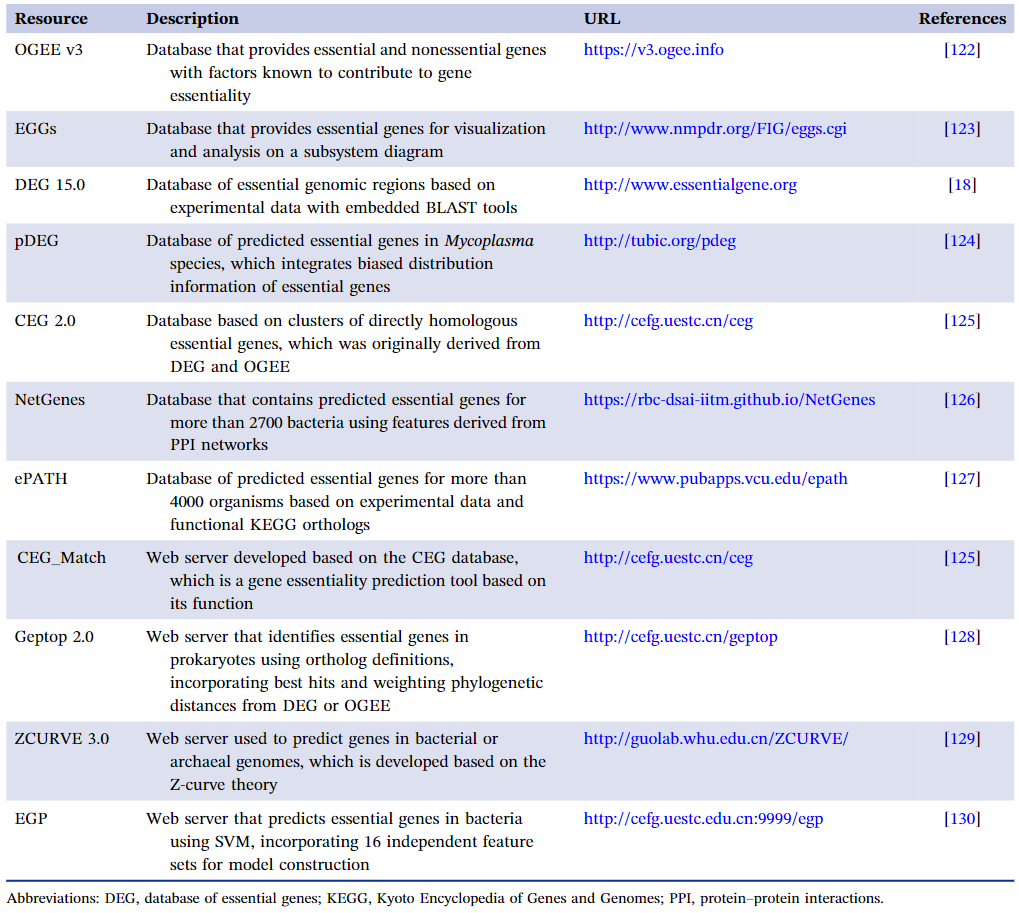
基于实验数据和计算模型,创建了必需基因的在线数据库等网络服务(表2)。研究人员可以使用这些数据库中的数据来研究必需基因/蛋白质的内在特性和与必需性密切相关的特征。除了基于实验得出的必需基因数据库外,还建立了存储预测的必需基因数据的数据库,以及一些开放访问程序,用于进行必需基因预测。
表2. 必需基因相关的数据库和网上服务
必需基因的实际应用
在合成生物学领域的应用
必需基因负责维持细胞中正常的生理和代谢过程,使它们对构建具有高稳定性或特定功能的细胞至关重要。因此,这些基因为基因组设计等相关研究提供了理论基础。目前,最小基因组是合成生物学中最重要的概念验证之一。最小基因组是指维持生物体最必需生命活动所需的基因组;它一直是合成生物学领域的一个关键研究目标。建立一个维持生命所需的最小通用基因集可以极大地增强我们对生命最必需层面的理解,并在生产中具有实际应用价值。作为合成生物学的基石,必需基因可以作为构建最小基因组的参考。值得注意的是,必需基因和最小基因组的概念并不完全等同。必需基因代表了成功繁殖所需的一组基因,而最小基因组代表了维持细胞存活所必需的基因。在实践中,鉴定和研究必需基因通常被用来推断最小基因组的组成。然而,对细菌代谢网络的计算建模表明,最小基因组所需的基因数量比所有必需基因的总和还要多。为了构建最小基因组,提出了自上而下的方法和自下而上的方法。自上而下的方法通过删除随机选定或未识别的基因组片段来减小基因组大小。删除可以通过各种实验方法完成,如基于质粒或基于线性DNA方法,以及利用特定位点的重组酶、转座子和CRISPR/Cas9系统。通过逐步移除非必需基因和功能元件的自上而下方法,已经实现了几个基因组的最小化。此外,研究人员还提出了一种名为MinGenome的自上而下基因组删减算法,该算法从最长可能的删减开始,连续删除代谢和调控基因。为了避免致命或生长缺陷相关基因的删除,该算法通过对生物质产量施加限制来保留必需基因和合成致死对。自上而下方法的主要优点是从一个可操作的基因组开始,允许通过恢复到最后一个未删减状态来补救由删除引起的任何不利影响。然而,这种方法耗时且可能导致意外的死胡同,因为在整个过程的每一步中,遗传景观都在改变,影响着其他基因的必需性。
自下而上的方法将基因片段与特定功能联系起来。DNA合成、测序技术和基因组移植的进步使得合成具有复杂基因组的长DNA序列成为可能。该方法主要利用聚合酶链反应技术,使得重叠的短寡核苷酸池的组装成为可能,并为自下而上方法提供了技术基础。基于逐步减少基因组的几轮合理设计和随机突变,还有研究者构建了一个近似的最小细菌基因组JCVI-syn3.0,它比最初预测的个体必需基因集多出98个基因。这一观察可以归因于原始基因组中的非必需基因在基因组减少过程中的合成致死而变得必需或准必需。Breuer等人利用支原体和细菌积累的代谢数据,并将其应用于通量平衡分析模型,建立了JCVI-syn3.0代谢网络的计算模型,这可以更好地预测JCVI-syn3.0中的必需和准必需基因。在自下而上设计过程中,有必要阐明每个基因及其在整个遗传背景中的相互作用的完整遗传信息。由于对基因组设计原理的了解有限以及目标生物体的复杂性,即使对于基因组较小的细菌,也存在众多可能的基因组配置,这对该方法构成了挑战。
正如上文所述,构建最小基因组的一个主要困难在于阐明整个遗传背景中基因之间的相互作用,这对必需基因的鉴定和基因组构建的早期阶段构成了重大挑战。然而,计算机辅助方法可以通过表征具有各种类型基因组修改的细胞,加速产生与遗传内容和细胞功能相对应的大规模数据,因此有可能拓宽和深化我们对整个细胞系统的理解,并有助于产生具有工业价值的生物系统。
在医学领域的应用
传统的药物和疫苗发现方法资源密集且耗时。最近,减法基因组学和反向疫苗学被归类为鉴定药物和疫苗候选目标的强大方法。这些方法通过消除昂贵且耗时的试错实验的需求,简化了药物开发。鉴定潜在靶标是药物和疫苗发现的第一步。考虑到必需基因的缺失或抑制可能对微生物产生致命效应,必需基因可以用作减法基因组学和反向疫苗学中药物和疫苗目标的筛选标准。此外,在癌症治疗领域,合成致死性的概念已扩展到成对基因,其中一个基因由于缺失或突变而失活,另一个基因的药物抑制导致癌细胞死亡,而正常细胞不受影响。在最直接的应用中,可以确定针对性治疗,杀死缺乏特定肿瘤抑制基因的癌细胞,但保留正常细胞。此外,癌细胞系中必需但在人体组织中非必需的基因可以揭示与相应癌症类型相关的致癌驱动因素、旁系基因表达模式和染色体结构。最近对大量癌细胞系进行的基因组规模CRISPR筛选的分析表明可能有数百个有效的药物靶标,但绝大多数是特定环境的。此外,一些必需基因已与人类疾病相关联。例如,通过基于人群分析的必需基因鉴定,人类大部分对功能丧失突变不耐受的基因属于必需基因,并且已发现它们在人类减数分裂重组中发挥作用,可能促进某些疾病的发生。此外,基因必需性的信息已被用于研究人类测序结果中与未知疾病相关的潜在致病基因变异。
然而,大多数现有的必需基因是在体外鉴定的,在体外和体内生长所需的基因之间可能存在显著差异。一个创新的解决方案是在非传统生长培养基中进行筛选,如在酸性培养皿中培育会造成肺部感染的铜绿假单胞菌。在筛选过程中优先考虑在营养有限培养基中表现活跃的产物对应的基因。在宿主模型中进行疾病筛选是另一种发现体内靶标的方法。例如,对被结核分枝杆菌感染的巨噬细胞进行高倍率显微镜筛选,鉴定了一系列针对细胞色素c的先导化合物。尽管对基因的系统研究揭示了众多潜在靶标,但在靶标验证过程中,通常需要对它们的功能有清晰的理解,这在鉴定药物靶标时构成了挑战。此外,药物抗性的持续进化是不可避免的,需要进一步理解抗性机制。最后,必须承认基因必需性是一种可进化的特性,必需性程度最大的基因可能是有前途的药物靶标。这在寻找药物靶标时带来了挑战,但也带来了机遇。因此,需要更多的研究来全面定义基因在不同环境和时间尺度下的必需性,并进一步探索必需基因的遗传背景。
DEG数据库的介绍
随着必需基因数据量的不断增长,迫切需要将这些数据组织成一个数据库,以便于使用这些数据。因此,我们实验室于2004年建立了DEG数据库,并随着实验数据的积累不断更新。该数据库汇编了从多种实验方法获得的全基因组必需基因数据。最新版本的数据库DEG 15.0,涵盖了细菌、真核生物和古菌,于2021年发布(https://tubic.org/deg)。总的来说,在DEG 15.0中存储了由不同实验方法确定的78组细菌、35组真核生物和2组古菌的必需基因数据集(图2A和2B),并展示了每组必需基因的具体信息(图2C)。此外,DEG数据库还包括与必需基因相关的分析模块,特别是针对细菌的,其中包含以下功能:(i)必需基因在先导链或后随链上的分布;(ii)必需基因的亚细胞定位分布(图2E);(iii)必需基因的同源群、EC号以及KEGG和GO富集信息(图2F,图2G);(ⅳ)定制的BLAST搜索工具,允许用户进行特定物种和实验的搜索,以识别已注释或未注释基因组中的必需基因(图2D)。除了必需基因外,该数据库还收集了一部分非必需基因的实验结果,以及除了编码蛋白质的必需遗传元件之外的内容,如非编码RNA和复制起始点。
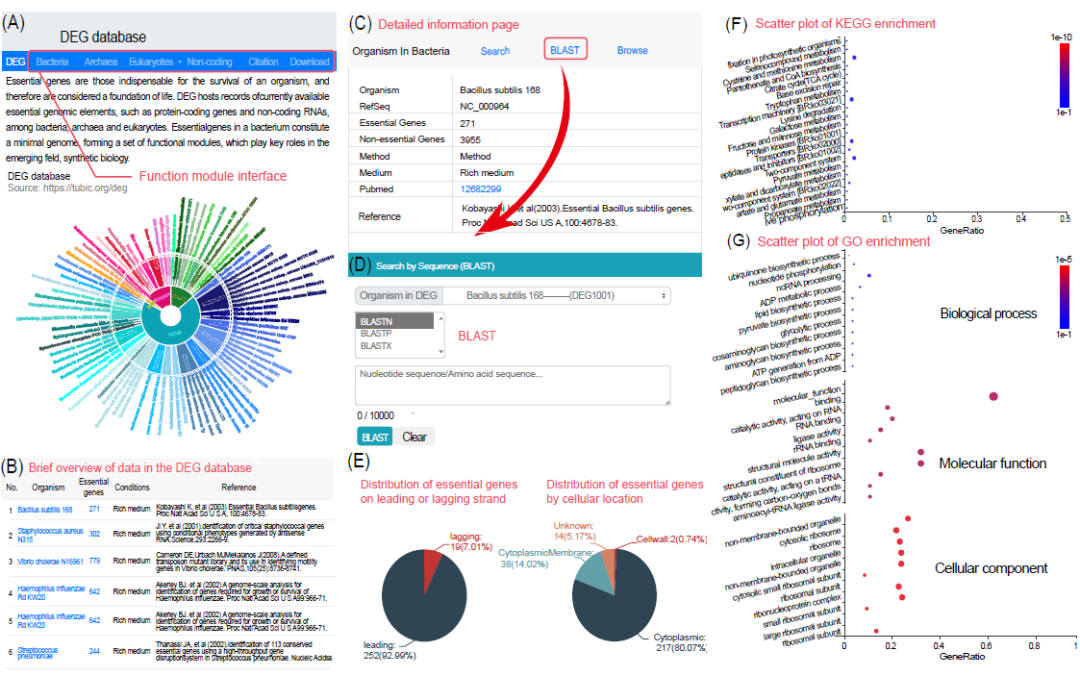
图 2. DEG数据库的概况
(A) DEG数据库的主页。提供了指向其他模块的接口,以及链接到不同物种中多个必需基因的实验结果信息。(B) DEG数据库中所有数据的汇总页面。(C) 特定菌株的实验结果相关信息截图,包括菌株信息、培养条件和参考文献引用。(D) 相应菌株的BLAST搜索界面。(E) 相应菌株中先导链/后随链上必需基因的分布。(F) 相应菌株中必需基因的京都基因与基因组百科全书(KEGG)富集分析结果。(G) 相应菌株中必需基因的基因本体(GO)富集分析结果。
DEG数据库的应用
DEG数据库已成为与必需基因相关研究的重要参考资料,是Nucleic Acids Research数据库专刊中最受欢迎的数据库(Golden Database)之一。使用DEG数据库可以快速访问特定物种的必需基因信息。目前,DEG数据库的应用主要集中在以下四个领域:人工基因组设计和构建、药物和疫苗设计、必需基因特征分析,以及必需基因的预测(图3)。
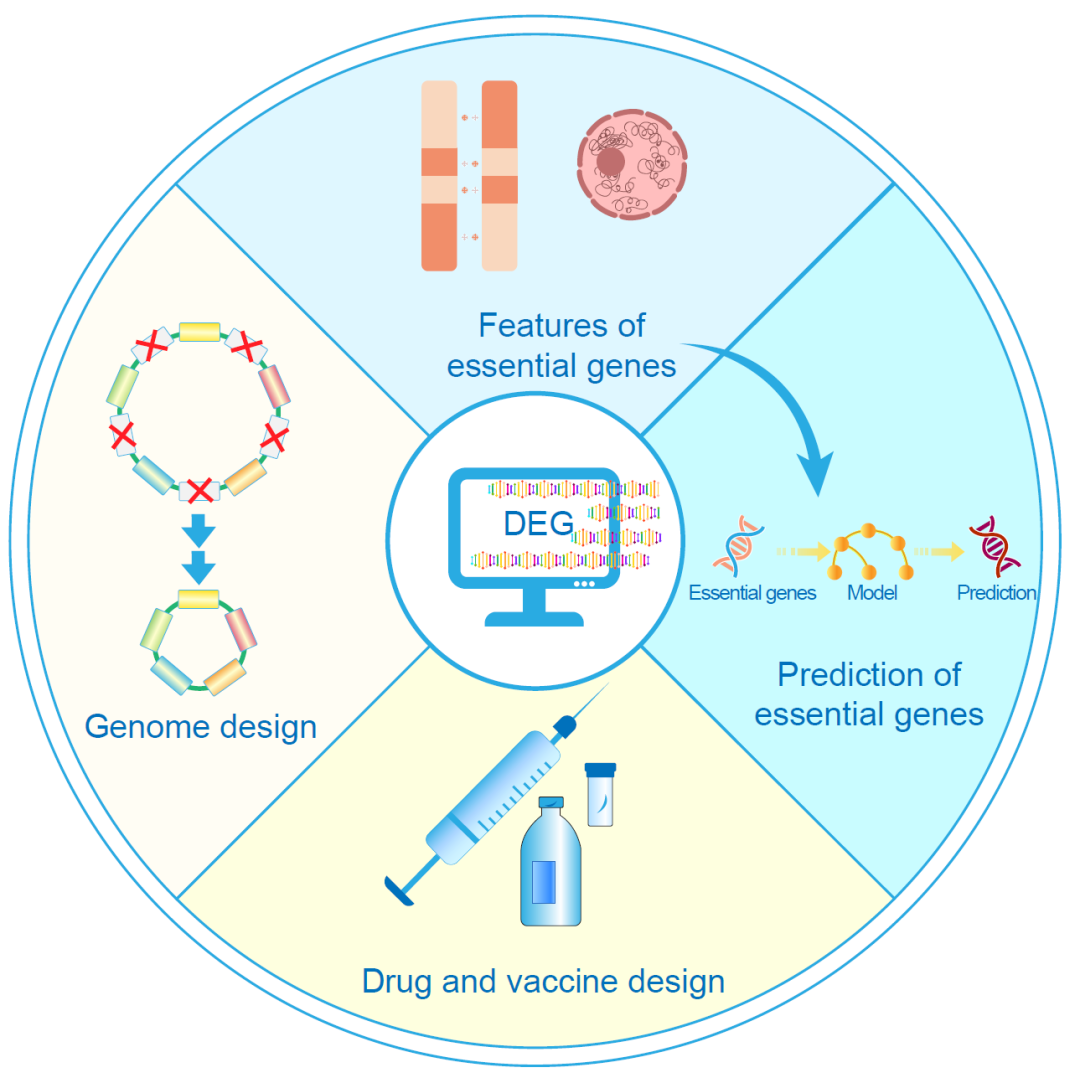
图 3. DEG数据库的主要应用
DEG数据库的应用主要集中在以下四个领域:人工基因组设计与构建、药物和疫苗设计、必需基因特性分析,以及必需基因的预测。
使用DEG数据库中的必需基因数据进行基因组设计的研究通常是自上而下的方法,因为基因组的非必需区域通常被视为待删除的候选区域。在自上而下的方法中,通常使用数据库中的必需/非必需基因数据来识别基因组中的删除位置,以提高其稳定性。一般来说,包含非必需基因的区域是删除的目标,而包含必需基因的区域通常被保留。此外,根据研究目的,还设计了删除运输蛋白基因、插入序列(ISs)、毒素-抗毒素对和其他功能元件。例如,刘等人选择性地从伯克霍尔德氏菌株中删除基因,以优化其基因组结构和生长速率。这项研究使用DEG数据库中的非必需基因数据来促进高度保守的基因组缩减菌株的鉴定,增强了基因组工程策略的可预测性,并提高了菌株生产的效率和稳定性。实际上,DEG数据库的数据也可为自下而上的基因组构建提供宝贵的参考。因此,DEG数据库被推荐为研究细菌存活所需最小基因组的参考资源。
使用计算策略而非培养微生物来识别潜在药物靶标可以显著降低时间和成本。DEG数据库中的基因信息已被用于鉴定病原细菌的必需蛋白。到目前为止,这种方法已被应用于确定包括假结核耶尔森菌、大肠杆菌、铜绿假单胞菌、索恩氏沙门菌、粪肠球菌、肺炎链球菌和假结核棒状杆菌在内的病原体的潜在药物靶标。此外,这一过程也适用于疫苗设计,称为反向疫苗学。反向疫苗学可以显著加速疫苗开发,因为它可以减少对单个抗原的广泛经验测试的需求。使用这种策略,已经鉴定了幽门螺杆菌、布鲁氏菌病、伤寒沙门和金黄色葡萄球的潜在疫苗靶标。此外,疫苗设计全流程管道VacSol和PanR也整合了DEG数据库中的数据用于辅助疫苗的设计。
DEG数据库提供了必需基因特征分析的宝贵数据,从而有助于必需基因特征领域的研究。数据库中包括非必需基因数据,使得可以进行两类基因之间的比较研究。已有许多研究利用DEG数据库提供的必需基因特征。例如,罗等人对细菌基因组进行了进化保守性分析,发现必需基因比非必需基因进化得更慢。必需基因在分布上的显著性链偏差主要与功能性有关,与基本生物功能相关(如翻译、转录和复制)的必需基因更偏向位于前导链上。令人惊讶的是,如果一个蛋白的特定功能域在多个生物中存在,则其必需性的可能性增加。此外,许多微生物的代谢网络已根据必需基因重建,并在一定程度上使用自动化系统如SEED和BiGG模型进行管理。基于DEG数据库和其他来源的数据,Magnúsdóttir等人系统分析了肠道微生物群的代谢相互作用以及外部因素对这些相互作用的影响。
在使用机器学习方法预测必需基因方面,DEG数据库提供了一个理想的训练数据集。例如,郭等人使用支持向量机(SVM)根据核苷酸序列数据派生的Z曲线特征来预测人类基因。曾等人构建了一个基于深度学习的框架,利用蛋白质-蛋白质相互作用(PPI)网络、基因表达数据和亚细胞定位信息预测必需基因。DeepHE通过整合序列数据和PPI网络的特征来预测人类的必需基因。施等人提出了一种名为iEsGene-CSMOTE的机器学习方法,该方法基于支持向量机,用于识别必需基因,引入了一种基于聚类的过采样技术CSMOTE,来克服数据不平衡的问题。除了利用必需基因特征外,周等人还提出了一种基于图像识别预测必需基因的算法,该算法使用了一种带有R-STDP学习规则的卷积脉冲神经网络,通过识别基于混沌游戏表示得到的DEG数据库中必需和非必需基因图像特征来预测必需基因。
基于DEG数据库的分析
必需基因与非必需基因的比较
由于DEG数据库15.0包含了大量已进行全基因组基因必需性筛选的细菌的必需基因和非必需基因数据集,因此它提供了探索基因组大小与必需基因数量之间相关性的可能。某些必需基因,如那些涉及复制、转录和翻译的基因,编码了所有基因组无论其大小如何都必需的必需细胞功能。因此,我们对DEG数据库中包含必需和非必需基因的数据进行了统计分析。线性回归结果显示,非必需基因数量与基因组大小呈正相关,而必需基因数量相对恒定(图4A)。无论基因组大小和实验条件如何,没有任何细菌物种的必需基因超过1000个。相对于基因总数,必需基因的百分比随着基因总数的增加而降低(图4B)。这一结果表明,必需基因的数量相对恒定,不随基因组长度变化,而非必需基因的数量与基因组长度呈正相关。
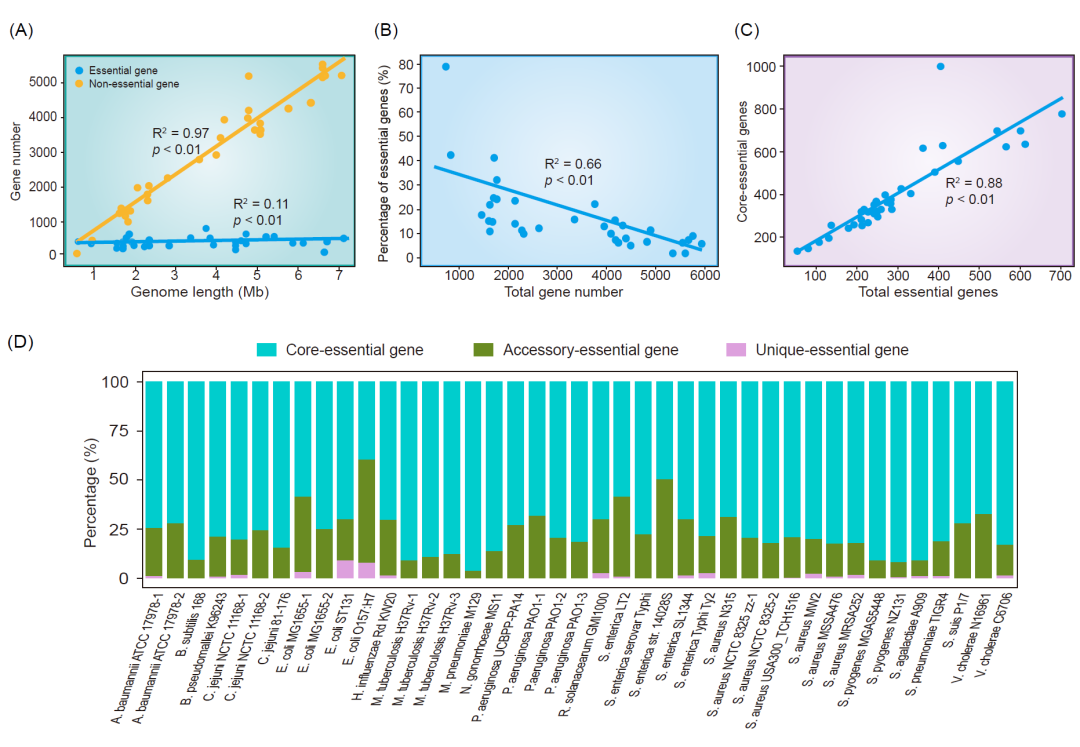
图 4. 基于DEG数据库中数据的统计分析
(A)基因组长度对应的必需基因和非必需基因数量。(B) 必需基因占总基因数量的百分比。(C) 核心必需基因数量与总必需基因数量之间的关系。(D) 全基因组框架下必需基因的分布。横轴代表不同物种。特异、附属和核心基因集中存在的必需基因分别用粉色、绿色和蓝色突出显示。纵轴代表不同类型必需基因的比例。
随后,我们对DEG数据库中必需和非必需基因的COG注释结果进行了统计分析。在C类(能量生产和转换)、F类(核苷酸转运和代谢)、H类(辅酶转运和代谢)、J类(翻译、核糖体结构和生物合成)、K类(转录)和O类(翻译后修饰、蛋白质转换和伴侣蛋白)中,必需基因的数量多于非必需基因。然而,在G类(碳水化合物转运和代谢)、N类(细胞运动)、M类(细胞壁、膜、被膜的生物合成)和U类(细胞内转运、分泌和囊泡运输)中,非必需基因的数量多于必需基。总之,COG功能富集分析表明,必需基因倾向于聚集在必需生命过程中,而非必需基因在环境适应和物质合成中扮演多样化的角色和功能。
泛基因组分类中必需基因的分布
“泛基因组”这一概念最早Tettelin在2008年提出,指的是一个物种的所有基因的集合,可以分为“核心基因”(所有菌株共有)、“附属基因”(两个或更多菌株共有)和“特异基因”(特定菌株独有)。构建一个物种的泛基因组能够突破单一参考基因组限制,使得在物种层面上对基因组进行研究成为可能。泛基因组分类中的核心基因是物种内所有个体共有的基因,通常执行重要功能,编码必需的生物学和表型相关性。考虑到必需基因和核心基因在生物体中的重要性,泛基因组分类已被纳入当前必需基因研究中。例如, Saxena等人成功鉴定了使用SRB(硫酸盐还原菌)模型的Oleidesulfovibrio alaskensis G20的关键必需基因集,并对这些基因进行了泛基因组分类。结果表明,大多数必需基因属于核心基因类别,而其他类别的必需基因可能是特定于环境的。此外,乌等人提出了一种策略,旨在减少枯草芽孢杆菌的基因组大小,同时确保保留核心和必需基因。
然而,目前缺乏大规模的泛基因组分类,特别是针对不同物种中必需基因的分类,以估计必需基因和核心基因的重叠程度。因此,我们提出了一种使用泛基因组框架对必需基因进行分类的方法。我们从NCBI下载了DEG数据库中包含物种的所有完整细菌菌株。为了确保泛基因组分析的准确性,我们选择了具有70个以上完整基因组的17个物种进行后续分析。首先,我们过滤N(未知)核苷酸超过1%的序列。接下来,我们排除了同一生物体的平均核苷酸同一性(ANI)值小于95%的菌株。ANI指的是两个微生物基因组中同源片段的平均碱基相似度。同一物种内的生物体之间的ANI值通常≥95%。最终,我们获得了与17个细菌物种相关的5900个菌株进行泛基因组分析。我们进行了泛基因组分析,并获得了17个物种的泛基因组(包括核心、附属和特异基因)的结果。通过将结果与DEG数据库中相应菌株的必需基因数据使用BLAST进行比较,我们确定了泛基因组分类中必需基因的分布(图4D)。
结果显示,除了大肠杆菌O157:H7外,细菌中的大多数必需基因属于核心基因。这表明,发挥重要作用的必需基因和核心基因在很大程度上重叠;这些类型的基因通常更加保守,被称为核心必需基因。线性回归分析显示,核心必需基因的数量与必需基因总数之间存在显著相关性(表4C)。然而,一些必需基因并不属于核心基因,这可能与菌株对特定生长条件的适应有关。因此,这种分类将为必需基因的功能提供新的见解。此外,将泛基因组分析与必需基因研究相结合为疫苗和药物的开发提供了新的视角。由于核心必需基因在更大程度上决定了病原体的生物学特征,识别核心必需基因可能有助于设计有效的广谱药物。相比之下,物种特异的必需基因可能是特定菌株药物的潜在靶标。
结论和未来展望
实验技术的进步促进了大规模全基因组水平筛选必需基因的实现,为研究许多生物过程中涉及的必需基因提供了宝贵的见解,揭示了必需基因的复杂和多面性,为它们在合成生物学和医学中的应用开辟了可能性。大量数据的产生也使得相关在线服务应运而生,例如相关数据库和工具的出现,它们为必需基因研究提供重要参考与帮助。然而,正如本文所讨论的,必需基因不是一个绝对概念,它取决于特定的环境和背景。由于分子相互作用的复杂性,使用计算方法,特别是机器学习方法来全面研究必需基因已被证明是具有发展前景的。在不久的将来,通过对具有不同理性设计基因组的细胞进行特征化,将加速产生有关遗传内容和细胞功能相关性的数据,这最终将拓宽我们对整个细胞系统的理解与必需基因在各个领域的应用。此外,我们也将致力于不断更新和维护DEG数据库。除了收集有关必需基因的实验数据外,我们还将关注必需基因的最新研究成果和新兴发展趋势。在未来,我们计划在以下几个领域增强和完善数据库:首先,我们旨在更有效地评估必需性的程度。在数据库的未来版本中,我们将添加通过实验鉴定的条件性必需基因注释来强调环境依赖性,将它们的必需性进一步基于在不同实验条件下存在的可能性进行分级。其次,我们还将探索为相应的必需基因提供跨物种链接的可能性,以便于在不同菌株或物种直接进行必需基因的特征比较。此外,我们还将为必需基因添加泛基因组分析模块,该模块会在泛基因组框架内将必需基因划分为核心、附属和特异三个类别,以更好地在物种水平上研究特定必需基因。最后,我们还将加入更多与必需性相关的特征分析信息,包括PPI(蛋白质-蛋白质相互作用)数据、表达谱和潜在药物靶标等,并考虑添加更多可视化手段以直观地呈现获得的结果。基于这一系列的更新,我们希望DEG数据库能够为必需基因研究提供更加全面、有价值的参考。
引文格式:
Ya-Ting Liang, Hao Luo, Yan Lin, Feng Gao. 2024. Recent advances in characterization of essential genes and development of a database of essential genes. iMeta e157. https://doi.org/10.1002/imt2.157
作者简介

梁雅婷(第一作者)
● 天津大学理学院物理系生物物理学专业硕士在读。
● 研究方向为必需基因与泛基因组研究,相关学术成果发表于iMeta等期刊。

高峰(通讯作者)
● 天津大学理学院教授、博导,天津大学生物信息中心主任。
● 主要从事微生物基因组生物信息学与合成生物学研究。在Nucleic Acids Research、PNAS、iMeta等国际知名刊物上发表第一/通讯作者SCI论文68篇(Nucleic Acids Research、Genomics Proteomics & Bioinformatics、Briefings in Bioinformatics和Bioinformatics 系列25篇),其中52篇为唯一第一/通讯作者,获Science、Nature等论文引用并佐证,相关成果得到中央电视台、《科技日报》、《人民日报》等国家级媒体报道。先后主持国家自然科学基金项目(5项)、国家重点研发计划课题(课题经费1091万)。
更多推荐
(▼ 点击跳转)
iMeta | 引用7000+,海普洛斯陈实富发布新版fastp,更快更好地处理FASTQ数据
iMeta | 德国国家肿瘤中心顾祖光发表复杂热图(ComplexHeatmap)可视化方法

1卷1期

1卷2期

1卷3期

1卷4期

2卷1期

2卷2期

2卷3期

2卷4期
期刊简介
“iMeta” 是由威立、肠菌分会和本领域数百位华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表原创研究、方法和综述以促进宏基因组学、微生物组和生物信息学发展。目标是发表前10%(IF > 15)的高影响力论文。期刊特色包括视频投稿、可重复分析、图片打磨、青年编委、50万用户的社交媒体宣传等。2022年2月正式创刊发行!目前期刊已经被ESCI、Scopus等数据库收录。
联系我们
iMeta主页:http://www.imeta.science
出版社:https://onlinelibrary.wiley.com/journal/2770596x
投稿:https://mc.manuscriptcentral.com/imeta
邮箱:office@imeta.science
高颜值免费 SCI 在线绘图(点击图片直达)
往期精品(点击图片直达文字对应教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
















































 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









