







该分析步骤来源于encode数据库,分析的数据也来源于encode数据库,分析流程基本是借用snakemake流程工具
第一步:进行原始数据的质控
该步骤使用软件fastqc(v0.11.9),环境为python3
rule fastqc:
input:
R1 = rawdata1+"/{sample}_R1.fq.gz",
R2 = rawdata1+"/{sample}_R2.fq.gz"
log:
R1 = rawdata+"/FASTQC/{sample}/{sample}_R1.log",
R2 = rawdata+"/FASTQC/{sample}/{sample}_R2.log",
output:
rawdata + "/FASTQC/{sample}/{sample}_R1_fastqc.html"
params:
rawdata+"/FASTQC/{sample}"
threads:60
shell:
"""
fastqc {input.R1} -o {params} -t {threads} > {log.R1} 2>&1
fastqc {input.R2} -o {params} -t {threads} > {log.R2} 2>&1
"""
第二步:鉴定数据中的UMI
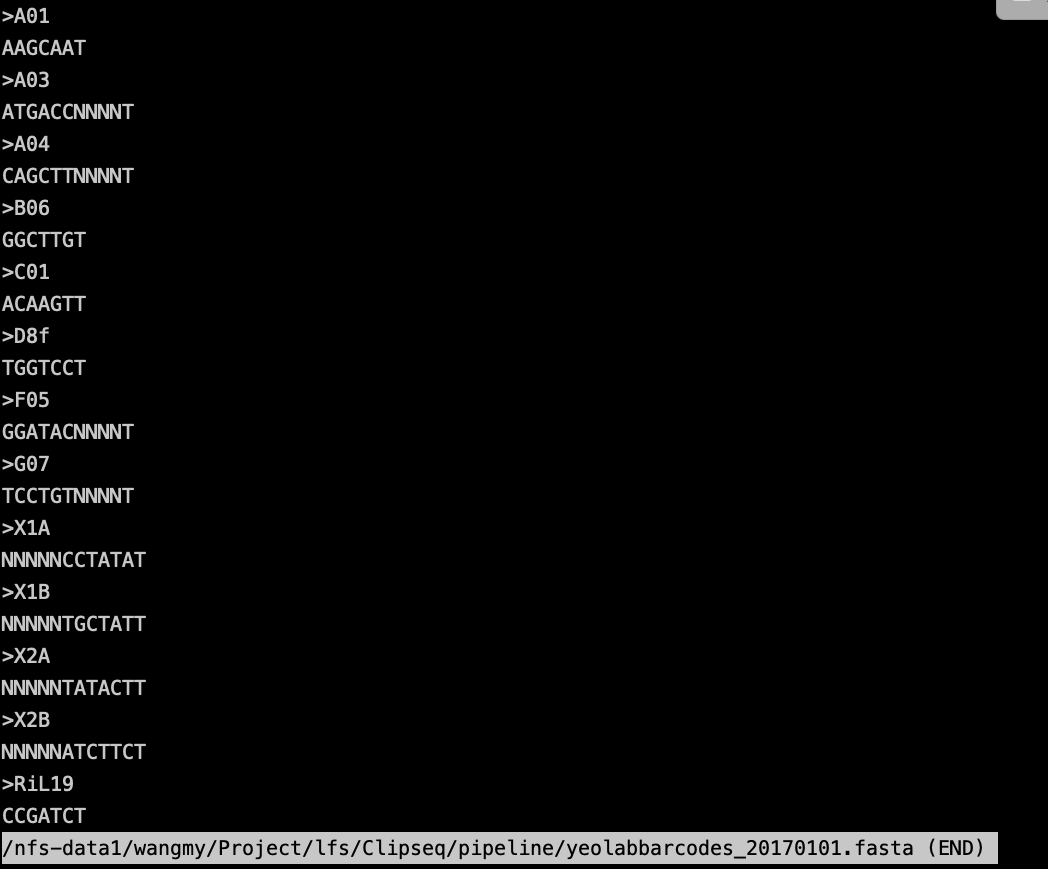
该步骤主要是对原始测序文件中的UMI进行鉴定,并且去除reads中的inline barcode。该步骤使用的程序是demux脚本,来源于Yeolab的eclipdemux。
该程序使用的环境应为python2,使用python3可能会报错。
rule Identify_UMI:
input:
R1 = rawdata1+"/{sample}_R1.fq.gz",
R2 = rawdata1+"/{sample}_R2.fq.gz"
output:
umi = rawdata+"/UMI/{sample}/{sample}.metrics",
r1 = rawdata+"/UMI/{sample}/{sample}.PE.NIL.r1.fq.gz",
r2 = rawdata+"/UMI/{sample}/{sample}.PE.NIL.r2.fq.gz"
params:
dir1 = rawdata+"/UMI/{sample}/",
name = "../xxx/UMI/{sample}/{sample}",
name1 = "PE"
shell:
"""
mkdir -p {params.dir1}
/data/xiaolab/wangmy/miniconda3/envs/python2/bin/python2.7 \
/nfs-data1/wangmy/software/eclipdemux/eclipdemux_package/demux.py \
-m {output.umi} \
--expectedbarcodeida RiL19 \
--expectedbarcodeidb RiL19 \
--fastq_1 {input.R1} \
--fastq_2 {input.R2} \
--dataset {params.name} --newname {params.name1} \
--barcodesfile yeolabbarcodes_20170101.fasta \
--length 5
"""
encode会在说明文档里介绍这次实验用的是那个inline barcode。如下:

inline barcode 它在接头的5’端即测序引物那部分上,和DNA片段邻近,在测序的时候,加入引物,然后一边合成一边测序,于是在最后的序列中就会引入barcode。
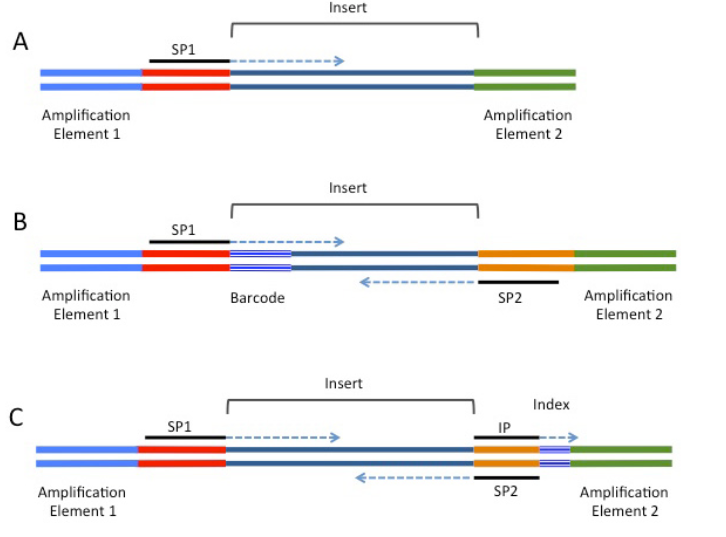
– barcodesfile 参数用于指定barcode序列和名字对应的关系,-m 是最后的umi对应文件。
第三步:去除接头并质控
该步骤总共去除两次接头,第一次去除read1和read2两端的接头。第二次去除read2的3ss端接头,目的是去除double ligation。每次去除过接头,还要质控看质量。
该步骤用的软件是cutadapt(4.3),环境为python3.
rule cutadapter1:
input:
R1 = rawdata+"/UMI/{sample}/{sample}.PE.NIL.r1.fq.gz",
R2 = rawdata+"/UMI/{sample}/{sample}.PE.NIL.r2.fq.gz"
output:
O= rawdata+"/CUTADAPT/{sample}/{sample}_R1_1.fq",
P= rawdata+"/CUTADAPT/{sample}/{sample}_R2_2.fq"
threads:50
shell:
"""
cutadapt \
--match-read-wildcards \
--times 1 \
-e 0.1 \
-O 1 \
--quality-cutoff 6 \
-m 18 \
--cores {threads} \
-a NNNNNAGATCGGAAGAGCACACGTCTGAACTCCAGTCAC \
-g CTTCCGATCTACAAGTT \
-g CTTCCGATCTTGGTCCT \
-A AACTTGTAGATCGGA \
-A AGGACCAAGATCGGA \
-A ACTTGTAGATCGGAA \
-A GGACCAAGATCGGAA \
-A CTTGTAGATCGGAAG \
-A GACCAAGATCGGAAG \
-A TTGTAGATCGGAAGA \
-A ACCAAGATCGGAAGA \
-A TGTAGATCGGAAGAG \
-A CCAAGATCGGAAGAG \
-A GTAGATCGGAAGAGC \
-A CAAGATCGGAAGAGC \
-A TAGATCGGAAGAGCG \
-A AAGATCGGAAGAGCG \
-A AGATCGGAAGAGCGT \
-A GATCGGAAGAGCGTC \
-A ATCGGAAGAGCGTCG \
-A TCGGAAGAGCGTCGT \
-A CGGAAGAGCGTCGTG \
-A GGAAGAGCGTCGTGT \
-o {output.O} \
-p {output.P} \
{input}
"""
#QC
rule cutadapter2:
input:
rawdata+"/CUTADAPT/{sample}/{sample}_R1_1.fq",
rawdata+"/CUTADAPT/{sample}/{sample}_R2_2.fq"
output:
O= rawdata+"/CUTADAPT/{sample}/{sample}_tr2_R1_1.fq",
P= rawdata+"/CUTADAPT/{sample}/{sample}_tr2_R2_2.fq"
threads:50
shell:
"""
cutadapt \
--match-read-wildcards \
--times 1 \
-e 0.1 \
-O 5 \
--quality-cutoff 6 \
-m 18 \
--cores {threads} \
-A AACTTGTAGATCGGA \
-A AGGACCAAGATCGGA \
-A ACTTGTAGATCGGAA \
-A GGACCAAGATCGGAA \
-A CTTGTAGATCGGAAG \
-A GACCAAGATCGGAAG \
-A TTGTAGATCGGAAGA \
-A ACCAAGATCGGAAGA \
-A TGTAGATCGGAAGAG \
-A CCAAGATCGGAAGAG \
-A GTAGATCGGAAGAGC \
-A CAAGATCGGAAGAGC \
-A TAGATCGGAAGAGCG \
-A AAGATCGGAAGAGCG \
-A AGATCGGAAGAGCGT \
-A GATCGGAAGAGCGTC \
-A ATCGGAAGAGCGTCG \
-A TCGGAAGAGCGTCGT \
-A CGGAAGAGCGTCGTG \
-A GGAAGAGCGTCGTGT \
-o {output.O} \
-p {output.P} \
{input}
"""
#再次QC
第四步:去除rRNA
该步骤主要是去除测序数据中的rRNA和重复序列reads,可以按照encode分析步骤。这里我自己构建的rRNA的index,利用bowtie2(2.4.5)
将比对到rRNA上的序列去除。
rule Remove_rRNA_Bowtie2:
input:
r1 = rawdata+"/CUTADAPT/{sample}/{sample}_tr2_R1_1.fq",
r2 = rawdata+"/CUTADAPT/{sample}/{sample}_tr2_R2_2.fq"
output:
sam = temp(rawdata + "/Mapping/Remove_rRNA/{sample}/{sample}.sam"),
O = temp(rawdata + "/Mapping/Remove_rRNA/{sample}/un-conc-mate.1"),
P = temp(rawdata + "/Mapping/Remove_rRNA/{sample}/un-conc-mate.2")
params:
idx = "~/database/NCBI/INDEX/rRNA_bowtie2/rRNA",
fqPath = rawdata+"/Mapping/Remove_rRNA/{sample}",
log:
rawdata+"/Mapping/Remove_rRNA/{sample}/{sample}.log"
threads:50
shell:
"""
bowtie2 -x {params.idx} -1 {input.r1} -2 {input.r2} -p {threads} -S {output.sam} --un-conc {params.fqPath} 1>{log} 2>&1
"""
第五步:mapping
该步骤使用STAR(2.7.9a),将上一步剩余的序列比对到人类hg38的基因组上。参数配置参见encode。
rule mapping:
input:
R1=rawdata+"/Mapping/Remove_rRNA/{sample}/un-conc-mate.1",
R2=rawdata+"/Mapping/Remove_rRNA/{sample}/un-conc-mate.2"
output:
rawdata+"/Mapping/{sample}/{sample}_Aligned.out.bam"
params:
Index="~/database/index/HG38",
#Index="~/database/index/HG19/STAR-79a",
Map=rawdata+"/Mapping/{sample}/{sample}_",
Gtf="~/database/hg38/gencode.v43.chr_patch_hapl_scaff.annotation.gtf"
threads:60
shell:
"""
STAR --runMode alignReads --runThreadN {threads} \
--genomeDir {params.Index} \
--genomeLoad NoSharedMemory \
--readFilesIn {input.R1} {input.R2} \
--outSAMunmapped Within --outFilterMultimapNmax 1\
--outFilterMultimapScoreRange 1 \
--outFilterScoreMin 10 \
--outFileNamePrefix {params.Map} --outSAMattrRGline ID:foo --outStd Log \
--outSAMtype BAM Unsorted --outFilterType BySJout --outSAMattributes All \
--outBAMcompression 10 --outSAMmode Full \
--outReadsUnmapped Fastx --alignEndsType EndToEnd
"""
第六步:去除pcr重复并提取reads
该步骤利用yeolab写的barcodecollapsepe.py程序进行去除pcr重复,该脚本来自eCLIP pipeline,也需要python2的操作环境。
提示:该步骤需要利用序列名字对bam文件进行排序,否则会报错;上一步mapping过程也需要–outSAMunmapped Within,否则也会
因为reads不成对而报错
测试数据使用的是[[RNAseq-链特异性#^5a533a|read2链特异性建库]],这里只选用read2作为后续分析。
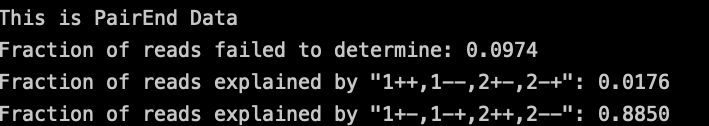
rule rmDup:
input:
rawdata+"/Mapping/{sample}/{sample}_Aligned.out.bam"
output:
rawdata+"/Mapping/{sample}/{sample}_rmDup.bam"
params:
rawdata+"/UMI/{sample}/{sample}.metrics"
threads:60
shell:
"""
samtools sort -n {input} -@ {threads} -o {input}
/data/xiaolab/wangmy/miniconda3/envs/python2/bin/python2 \
/nfs-data1/wangmy/software/eCLIP/bin/barcodecollapsepe.py \
-o {output} \
-m {params} \
-b {input}
samtools sort {output} -@ {threads} -o {output}
samtools index -@ {threads} -b {output}
"""
rule exR2:
input:
rawdata+"/Mapping/{sample}/{sample}_rmDup.bam"
output:
rawdata+"/Mapping/{sample}/{sample}_rmDup_r2.bam"
threads:60
shell:
"""
samtools view -f 128 -@ {threads} -b -o {output} {input}
samtools index -@ {threads} -b {output}
"""
第七步:可视化
该步骤使用的程序是来自于yeolab的makebigwigfiles,使用的是python2环境。生成正负链对应的bw文件,可在IGV中进行可视化。
#python2
NAME=(
)
for i in "${NAME[@]}";
do
mkdir -p ../U2AF2/BigWig/${i}
makebigwigfiles --bw_pos ../xxx/BigWig/${i}/${i}_rmDup_r2_pos.bw --bw_neg ../xxx/BigWig/${i}/${i}_rmDup_r2_neg.bw --bam ../xxx/Mapping/${i}/${i}_rmDup_r2.bam --genome ~/database/hg38/hg38.chrom.sizes --direction r
done
第八步:callpeak
该步骤使用yeolab的clipper工具,对clip数据进行callpeak。该软件可以在python3环境下运行。
rule fastqc:
input:
rawdata + "/Mapping/{sample}/{sample}_rmDup_r2.bam"
output:
rawdata + "/BigBed/{sample}/{sample}_rmDup_r2.bed"
threads:60
shell:
"""
clipper \
--species hg38 \
--bam {input} \
--save-pickle \
--outfile {output}
"""
第九步:Input normalization并融合两组重复数据。
这部分内容并没有完全看懂,只是跟着做了一遍。后续会在理解一下。
该步骤借鉴yeolab的merge_peaks,由于该流程中的各种安装包比较老,在使用的时候各种报错。因此我利用其中提供的perl程序,构建了我自己的snakemake流程。处理流程如下:

1. normalization
该步骤主要利用input对CLIP数据进行归一化,简单来说就是去除input也存在的peak(背景)。在归一化之前,还需要利用samtools计算样本的reads数。
# -c对比对上的reads进行计数,F跳过某个flag,f表示需要某个flag,q表示flag最小的值
# 数字4代表该序列没有比对到参考序列上
# 数字8代表该序列的mate序列没有比对到参考序列上
samtools view -cF 4 {input.IP} > {output.IP}
#提取input的reads数
rule input1:
input:
rawdata + "/Mapping/Control_HepG2_rep1/Control_HepG2_rep1_rmDup_r2.bam"
output:
rawdata + "/BigBed/Control_HepG2_rep1/Control_HepG2_rep1_rmDup_r2.txt"
shell:
"""
samtools view -cF 4 {input} > {output}
"""
#1.提取IP的reads数
#2.overlap_peakfi_with_bam_PE.pl 对IP进行归一化
#3.ompress_l2foldenrpeakfi_for_replicate_overlapping_bedformat_outputfull.pl 对peak进行合并
rule normalization:
input:
IP = rawdata + "/Mapping/{sample}/{sample}_rmDup_r2.bam",
controlTxT = rawdata + "/BigBed/Control_HepG2_rep1/Control_HepG2_rep1_rmDup_r2.txt"
output:
IP1 = rawdata + "/BigBed/{sample}/{sample}_rmDup_r2.txt",
norm = rawdata + "/BigBed/{sample}/{sample}_rmDup_r2_normalization.bed",
normF = rawdata + "/BigBed/{sample}/{sample}_rmDup_r2_normalization.compressed.bed",
normFF = rawdata + "/BigBed/{sample}/{sample}_rmDup_r2_normalization.compressed.bed.full"
params:
control = rawdata + "/Mapping/Control_HepG2_rep1/Control_HepG2_rep1_rmDup_r2.bam",
bed = rawdata + "/BigBed/{sample}/{sample}_rmDup_r2.bed",
norm = rawdata + "/BigBed/{sample}/{sample}_rmDup_r2_normalization.bed.full"
threads:60
shell:
"""
samtools view -cF 4 {input.IP} > {output.IP1}
perl /nfs-data1/wangmy/software/eCLIP/bin/overlap_peakfi_with_bam_PE.pl \
{input.IP} {params.control} {params.bed} \
{output.IP1} {input.controlTxT} \
{output.norm}
perl /nfs-data1/wangmy/software/merge_peaks/bin/perl/compress_l2foldenrpeakfi_for_replicate_ove
rlapping_bedformat_outputfull.pl \
{params.norm} {output.normF} {output.normFF}
"""
^59c555
2. 计算熵值,并转换成bed6格式
rule normalization1:
input:
normFF = rawdata + "/BigBed/{sample}/{sample}_rmDup_r2_normalization.compressed.bed.full"
output:
entropy = rawdata + "/BigBed/{sample}/{sample}_rmDup_r2_normalization.compressed.bed.full.entro
py",
excess = rawdata + "/BigBed/{sample}/{sample}_rmDup_r2_normalization.compressed.bed.full.excess
_reads",
entropy_bed6 = rawdata + "/BigBed/{sample}/{sample}_rmDup_r2_normalization.compressed.bed.full.
bed"
params:
controlTxT = rawdata + "/BigBed/Control_HepG2_rep1/Control_HepG2_rep1_rmDup_r2.txt",
IP1 = rawdata + "/BigBed/{sample}/{sample}_rmDup_r2.txt"
shell:
"""
perl /nfs-data1/wangmy/software/merge_peaks/bin/perl/make_informationcontent_from_peaks.pl \
{input.normFF} {params.IP1} {params.controlTxT} {output.entropy} {output.excess}
python /nfs-data1/wangmy/software/merge_peaks/bin/full_to_bed.py \
--input {output.entropy} --output {output.entropy_bed6}
"""
3. 利用IDR合并两组重复
rule IDR:
input:
entropy_bed6_rep1 = rawdata + "/BigBed/{sample}_rep1/{sample}_rep1_rmDup_r2_normalization.compressed.bed.full.bed",
entropy_bed6_rep2 = rawdata + "/BigBed/{sample}_rep2/{sample}_rep2_rmDup_r2_normalization.compressed.bed.full.bed"
output:
idr1 = rawdata + "/BigBed/{sample}/{sample}_rmDup_r2_normalization.compressed.idr.bed"
log:rawdata + "/BigBed/{sample}/{sample}_rmDup_r2_normalization.compressed.idr.log"
shell:
"""
/data/xiaolab/wangmy/miniconda3/envs/python35/bin/idr \
--samples {input.entropy_bed6_rep1} {input.entropy_bed6_rep2} \
--input-file-type bed \
--rank 5 \
--plot \
--peak-merge-method max \
--log-output-file {log} \
--output-file {output}
"""
使用idr2.0.4会报错,这里使用2.0.2版本(python3.5)。
如果使用中遇到ValueError: The truth value of an array with more than one element is ambiguous. Use a.any() or a.all()的报错。
可以试试把第415行的if localIDRs == None or IDRs == None:改成if localIDRs.all() == None or IDRs.all() == None:
并对合并后的peak进行统计:
rule IDR1:
input:
entropy_bed6_rep1 = rawdata + "/BigBed/{sample}_rep1/{sample}_rep1_rmDup_r2_normalization.compre
ssed.bed.full.entropy",
entropy_bed6_rep2 = rawdata + "/BigBed/{sample}_rep2/{sample}_rep2_rmDup_r2_normalization.compre
ssed.bed.full.entropy",
idr1 = rawdata + "/BigBed/{sample}/{sample}_rmDup_r2_normalization.compressed.idr.bed"
output:
rawdata + "/BigBed/{sample}/{sample}_rmDup_r2_normalization.compressed.idr.summary"
shell:
"""
perl /nfs-data1/wangmy/software/merge_peaks/bin/perl/parse_idr_peaks.pl \
{input.idr1} {input.entropy_bed6_rep1} {input.entropy_bed6_rep2} \
{output}
"""
4. 对idr合并后的peak再次归一化
参考[[eclip分析流程#^59c555|第一步]]
5. 合并peak
rule mergepeaks:
input:
full_rep1 = rawdata + "/BigBed/{sample}_rep1/{sample}_rep1_rmDup_r2_normalization.idr.bed.full",
full_rep2 = rawdata + "/BigBed/{sample}_rep2/{sample}_rep2_rmDup_r2_normalization.idr.bed.full"
output:
full_rep1 = rawdata + "/BigBed/{sample}_rep1/{sample}_rep1_rmDup_r2_normalization.idr.bed.full.o
utput",
full_rep2 = rawdata + "/BigBed/{sample}_rep2/{sample}_rep2_rmDup_r2_normalization.idr.bed.full.o
utput",
bed6= rawdata + "/BigBed/{sample}/{sample}_rmDup_r2_normalization.idr.bed.full.bed",
bed9= rawdata + "/BigBed/{sample}/{sample}_rmDup_r2_normalization.idr.bed.full.custombed.bed"
params:
entropy_bed6_rep1 = rawdata + "/BigBed/{sample}_rep1/{sample}_rep1_rmDup_r2_normalization.compre
ssed.bed.full.entropy",
entropy_bed6_rep2 = rawdata + "/BigBed/{sample}_rep2/{sample}_rep2_rmDup_r2_normalization.compre
ssed.bed.full.entropy",
idr1 = rawdata + "/BigBed/{sample}/{sample}_rmDup_r2_normalization.compressed.idr.bed"
threads:60
shell:
"""
perl /nfs-data1/wangmy/software/merge_peaks/bin/perl/get_reproducing_peaks.pl \
{input.full_rep1} {input.full_rep2} \
{output.full_rep1} {output.full_rep2} \
{output.bed6} {output.bed9} \
{params.entropy_bed6_rep1} {params.entropy_bed6_rep2} \
{params.idr1}
"""
在使用过程中,或许你需要安装perl的模块
cpan install Statistics::Basic
cpan install Statistics::Distributions
cpan install Statistics::R
#使用perl程序需要安装这几个模块,如果安装不了,可以直接下载包安装
#从 CPAN(https://metacpan.org/)下载模块
wget https://cpan.metacpan.org/authors/id/F/FA/FANGLY/Statistics-R-0.34.tar.gz
#解压并进入
tar -xvf Statistics-R-0.34.tar.gz
cd Statistics-R-0.34.tar.gz
#生成makefile文件
perl Makefile.PL
#生成模块
make
#安装模块
make install
#查看已安装的包
perldoc perllocal

















 474
474

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









