







小罗碎碎念
顶刊速递|AI+免疫组化
今天分享的文献主题是——人工智能与免疫组化的结合。其实之前的推文中也偶尔会提到免疫组化相关的内容,但是一直没有出一期,系统的梳理一下这一部分内容在医学AI中的应用,所以这期推文便来完成一半的任务。
为什么说是完成一半的任务呢?因为目前文献中,免疫组化的应用可以大致分为两类——验证&简化。所谓验证,是指我先根据已有的数据训练模型,然后再用少量的免疫组化数据来验证我的模型,今天介绍的六篇推文都是这种套路。
第二类应用是简化,这个稍微复杂点。我们都知道免疫组化其实流程是有些繁琐的,并且耗时,搞完一批样本周期还是挺长的,所以所谓的简化就是指通过训练我们提出的模型,在普通的病理切片上实现免疫组化想实现的效果,这就会联系到另一块的应用——虚拟染色以及染色标准化。你看,这还能病理切片的预处理流程联系起来,是不是很有意思。
好!点到为止,我是罗小罗同学,明天也要记得来看小罗的推文哦!!

一、从多模态到单模态的知识蒸馏,实现淋巴瘤亚型的分类任务

文献概述
这篇文章提出了一个基于视觉变换器(Vision Transformer)的框架,用于从多模态到单模态的知识转移,以对淋巴瘤亚型进行分类。
研究团队指出,确定淋巴瘤亚型对于改善患者治疗和提高生存率至关重要。目前,依赖基因表达技术的金标准诊断方法成本高昂且耗时,而基于IHC(免疫组化)技术的替代诊断方法虽然被世界卫生组织推荐,但准确性仍有待提高。
为了解决这些问题,作者提出了一种使用深度学习模型分析全切片图像(Whole Slide Images, WSI)的方法,这提供成本效益高且快速的替代方案。他们开发了一个多模态架构,用于从多种WSI模态中训练分类模型,然后通过知识蒸馏过程引导单模态分类器的学习。
在157名淋巴瘤患者的数据集上进行的实验表明,单模态分类模型的性能超过了六种最新的最先进方法。此外,通过实验数据估计的幂律曲线表明,通过合理数量的额外患者训练数据,该模型的诊断准确性可与IHC技术相媲美。该框架的效率也通过在乳腺癌数据集(BCI数据集)上的额外实验得到了证实。
重点关注
图1展示了一个从多模态到单模态的知识蒸馏(Knowledge Distillation, KD)过程,用于WSI亚型分类模型。

这个过程包括两个阶段:
-
第一阶段 - 教师模型训练(Multi-modal teacher model training):
- 在这个阶段,教师模型被训练以识别来自HES(Hematoxylin-Eosin-Safran)和IHC(Immunohistochemistry)模态的WSI。
- 教师模型使用多模态输入,即同时使用HES和IHC标记的图像,来训练分类器。
-
第二阶段 - 学生模型训练(Student model training):
- 在这个阶段,教师模型的知识通过知识蒸馏过程传递给学生模型,学生模型仅使用HES模态的WSI进行训练。
- 学生模型的目的是学习如何仅依赖单一模态(HES)来区分淋巴瘤的亚型。
图1中还展示了以下元素:
- WSI HES & WSI IHC:分别代表HES和IHC模态的全切片图像。
- Trained model:表示训练好的模型,这里指的是教师模型。
- Mono-modal student model:单模态学生模型,它在训练过程中只使用HES模态。
- Knowledge Distillation:知识蒸馏过程,通过这个过程,教师模型的预测结果被用作学生模型训练的一部分。
- Gold Standard diagnosis:金标准诊断,这里作为评估模型性能的标准。
- Teacher loss & Student loss:分别代表教师模型和学生模型的损失函数。
- β:损失函数中的一个超参数,用于平衡教师模型预测和实际目标之间的差异。
整个框架的目的是利用多模态教师模型学习到的丰富特征,通过知识蒸馏过程,指导单模态学生模型学习到区分淋巴瘤亚型的关键特征。这种方法可以减少对IHC标记的依赖,从而简化实验室的数据获取过程,加速患者诊断,并降低成本。
二、识别乳腺癌的精确生物标志物改善患者预后,并解决传统分期在预测治疗反应方面的局限性

文献概述
这篇文章是一项关
于乳腺癌的研究。
研究的主要目的是通过识别乳腺癌的精确生物标志物来改善患者预后,并解决传统分期在预测治疗反应方面的局限性。
研究方法包括:
- 分析了超过7000名乳腺癌患者的数据,涵盖了14个数据集,包括临床数据和8名患者的单细胞数据。
- 使用了
10种机器学习算法的54种独特组合来分析100个现有的乳腺癌标志物。 - 进行了
免疫组化实验以实证验证研究结果。 - 研究还探讨了潜在的免疫疗法和化疗方法。
研究结果:
- 确定了
五个与谷氨酰胺代谢重编程(GMR)相关的基因,形成了一个新的GMR模型的基础。 - 该模型在预测复发和死亡风险方面显示出比现有临床和分子特征更高的准确性。
- 被模型分类为高风险的患者预后较差。
- 在30名患者中进行的IHC验证加强了这些发现,表明该模型具有广泛的适用性。
- 模型还表明了不同的治疗方法反应:低风险患者从免疫疗法中受益更多,而高风险患者对特定化疗药物如BI-2536和ispinesib表现出敏感性。
结论:
- GMR模型在乳腺癌预后和治疗策略个性化方面迈出了重要一步,为有效管理不同乳腺癌患者群体提供了重要见解。
重点关注
研究的总体流程。

-
Data Acquisition(数据获取):研究从TCGA数据库和GEO数据库收集了乳腺癌患者的基因表达数据、突变数据和临床信息。确保了数据集的完整性和准确性。
-
Single-Cell Sequencing Technique(单细胞测序技术):使用单细胞数据,经过数据预处理,包括去除零表达基因,保留非零表达基因,并使用Seurat R包进行数据标准化和降维。
-
Cell-Type Annotation(细胞类型注释):通过特定的算法对细胞进行分类,以确定不同的细胞类型。
-
Cell-Cell Communication Analysis(细胞间通讯分析):使用CellChat工具分析细胞间的通讯模式,比较不同细胞群之间的相互作用。
-
Functional Analysis(功能分析):利用GO和KEGG数据库评估GMR相关基因在肿瘤和正常组织中的表达差异。
-
Calculating the GMR-Score(计算GMR得分):基于与谷氨酰胺代谢相关的基因表达差异,开发了一个GMR得分系统,用于评估肿瘤微环境中的代谢活动。
-
Construction of the GMR Model and Nomogram(构建GMR模型和列线图):使用多种计算算法,包括随机森林、LASSO、梯度提升机等,来构建一个基于GMR的预后模型,并开发了一个列线图来预测乳腺癌患者的生存概率。
-
Validation of the GMR Model(GMR模型的验证):通过Kaplan-Meier生存分析和Cox回归分析等方法,验证模型的普适性和可靠性。
-
Genomic Alteration Analysis(基因组变异分析):分析高风险和低风险乳腺癌亚组之间的遗传差异,包括突变频率和拷贝数变异。
-
Identifying TME Disparities(识别TME差异):评估GMR模型分类的患者中肿瘤微环境中免疫细胞的浸润水平。
-
Determining Therapeutic Targets and Drugs(确定治疗靶点和药物):基于药物重利用中心的数据,预测化疗反应并确定潜在的治疗靶点。
-
Immunohistochemistry Experiment(免疫组化实验):收集手术中的乳腺癌组织样本,进行Hematoxylin和Eosin染色,并由病理学家进行评估。
整个流程是一个综合性的分析,从数据收集到模型构建,再到验证和潜在治疗策略的探索,旨在提高乳腺癌的治疗效果和患者的生存率。
三、基于乙肝病毒开发肝细胞癌相关的分类和预后模型

文献概述
这篇文章是关于乙型肝炎病毒(HBV)相关肝细胞癌(HCC)的研究,特别是基于程序性细胞死亡(PCD)相关基因的分类和预后模型的开发。
- 研究背景:HBV感染是HCC的主要风险因素。PCD是抑制肿瘤生长的关键过程,PCD相关基因的变化促进HBV-HCC的发展。
- 研究目的:开发一个结合基因组和临床信息的预后模型,通过生物信息学分析和实验验证,提供HBV-HCC分子异质性的新见解。
- 方法:分析了
139个来自癌症基因组图谱(TCGA)的HBV-HCC样本,并使用基因表达综合数据库(GEO)的30个样本进行验证。使用了多种生物信息学工具,包括差异表达分析、基因集变异分析和机器学习算法,对HBV-HCC患者的RNA测序数据进行全面分析。特别关注了DLAT基因,并在组织芯片和患者队列中进行了进一步研究。 - 结果:聚类分析识别了三个不同的HBV-HCC患者亚组。其中,第二组在DNA复制相关途径和肿瘤相关过程中表现出显著激活。PCD相关基因的拷贝数变异(CNV)分析也揭示了三个亚组中的不同模式,与途径激活和生存结果的差异有关。HBV-HCC患者肿瘤组织中的DLAT表达上调。
- 讨论:基于PCD相关基因,开发了一个预后模型,提供了HBV-HCC分子异质性的新见解。研究强调了PCD相关基因,特别是DLAT的重要性,并在体外进行了研究,以探索其潜在的临床意义。
- 结论:通过研究,基于不同的PCD相关基因对HBV-HCC亚组进行分类,并构建了预后模型。研究表明PCD相关基因可以作为患者分层和个性化治疗的潜在生物标志物。
- 研究限制:样本量相对较小,需要更大样本量的外部验证来确认研究结果。需要进一步研究确定途径和PCD相关基因背后的分子机制。研究基于HBV-HCC肝组织的转录组数据,需要使用不同HBV-HCC样本的其他组学数据进行进一步验证。
重点关注
图1展示了基于基因表达谱对乙型肝炎病毒相关
肝细胞癌(HBV-HCC)样本进行的聚类分析。
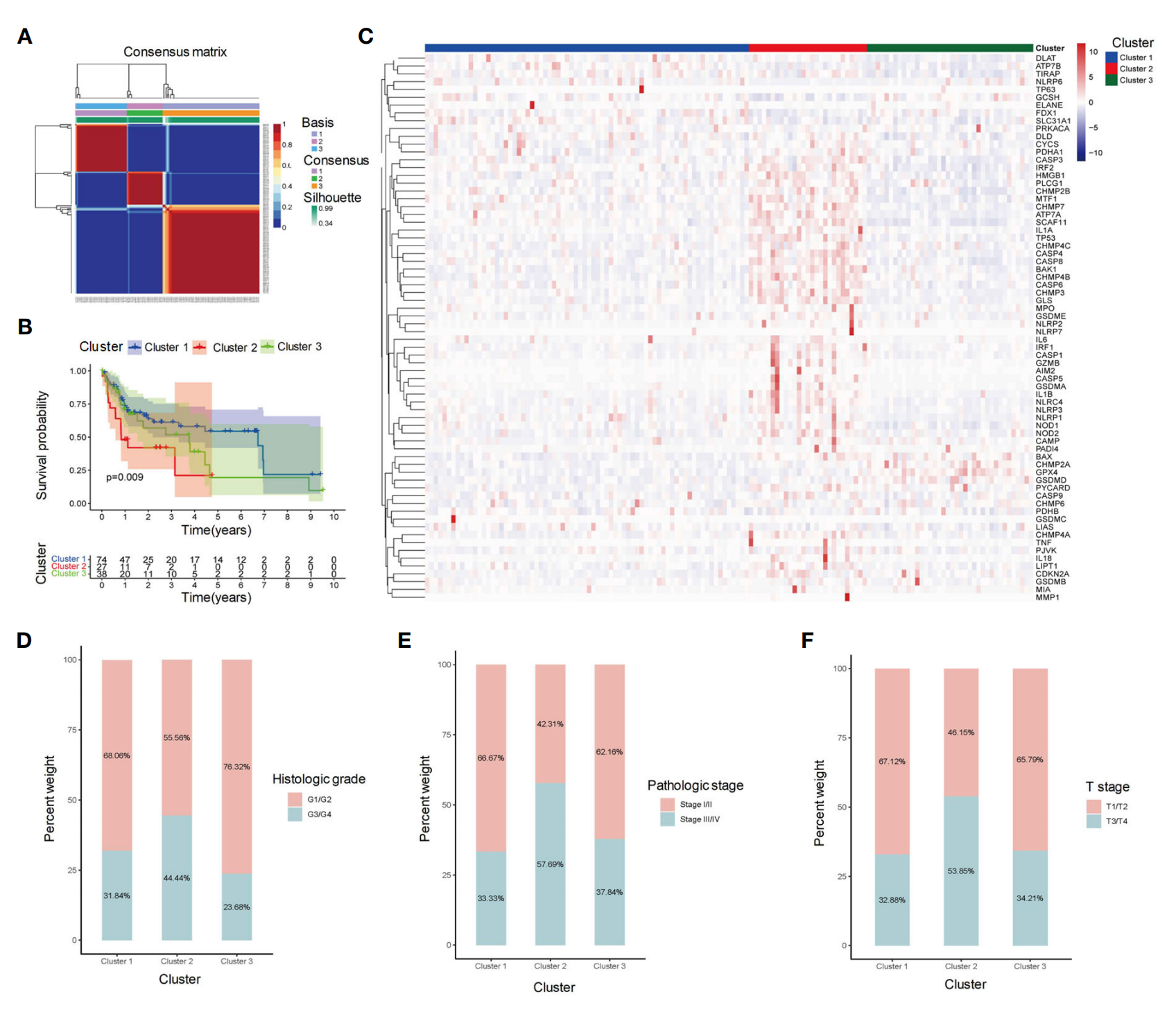
以下是对图1各部分的分析:
A部分(Clustering heatmap):
- 这部分的热图展示了通过非负矩阵分解(NMF)无监督聚类方法识别出的三个不同的HBV-HCC样本亚组。
- 这些亚组被标记为Cluster 1、Cluster 2和Cluster 3。
- 热图显示了不同样本间基因表达的差异,颜色的深浅代表了基因表达水平的高低。
B部分(Kaplan-Meier survival analysis):
- 这部分的Kaplan-Meier生存分析图显示了每个亚组患者的总体生存(OS)情况。
- 通过生存曲线,可以观察到不同亚组之间生存率的差异。
- 曲线的下降速度反映了生存率随时间的变化情况,曲线越低表示生存率越低。
C部分(Heatmap of PCD-related gene expression):
- 这部分的热图展示了与程序性细胞死亡(PCD)相关的基因在每个亚组中的表达水平。
- 热图中的颜色变化表示基因表达的相对强度,有助于识别哪些基因在特定亚组中表达上调或下调。
D-F部分(Stacked bar plot):
- 这些堆叠条形图展示了每个亚组中患者组织学分级、病理分期和T分期的分布情况。
- 通过这些图表,可以了解不同临床特征在各个亚组中的分布频率。
- 例如,如果某个亚组有更多G3/G4分级或III/IV分期的样本,这表明该亚组的患者具有更高的风险。
总体分析:
- 图1提供了HBV-HCC样本基于基因表达的聚类信息,以及与患者生存率和临床特征相关的数据。
- 通过这些分析,研究人员能够识别出具有不同临床结果和生存率的HBV-HCC患者亚组。
- 这些信息对于理解HBV-HCC的分子异质性、开发个性化治疗策略和预后评估具有重要意义。
四、单细胞测序数据,识别胃肠道癌症中的新抗原反应性CD8+ T细胞(Neo T细胞)

文献概述
这篇文章描述了一种基于单细胞测序数据的机器学习方法,用于识别胃肠道癌症中的新抗原反应性CD8+ T细胞(Neo T细胞)。
研究背景指出,Neo T细胞是患者对胃肠道癌症免疫反应的主要驱动力,但传统方法在识别这些细胞及其相应的T细胞受体(TCRs)方面非常耗时和复杂。
研究方法包括开发了一个包含26个基因的机器学习模型,通过分析数千个肿瘤浸润性淋巴细胞的单细胞转录组来识别新抗原反应性T细胞。
研究结果显示,该模型在训练和验证集上表现良好,发现Neo T细胞在与酰胺相关信号通路的生物学过程中存在显著差异。通过分析潜在的细胞间相互作用,结合空间转录组学和多重免疫组化数据,揭示了Neo T细胞具有强大的信号分子,包括LTA,这与肿瘤微环境中的肿瘤细胞相互作用,从而发挥抗肿瘤效应。
通过分析接受新辅助免疫疗法治疗的患者的肿瘤样本中的CD8+ T细胞,确定了Neo T细胞的比例与患者的临床益处和总体生存率显著正相关。与实验过程中识别Neo T细胞的传统方法相比,这种方法减少了实验平台的要求,缩短了识别周期和成本,并降低了临床应用ACT的门槛。这些发现鼓励进一步研究这种策略如何被用来开发更有效的癌症免疫疗法。
重点关注
图1展示了特征重要性分析,该分析识别了与新抗原反应性T细胞(Neo T细胞)强相关的26个基因。
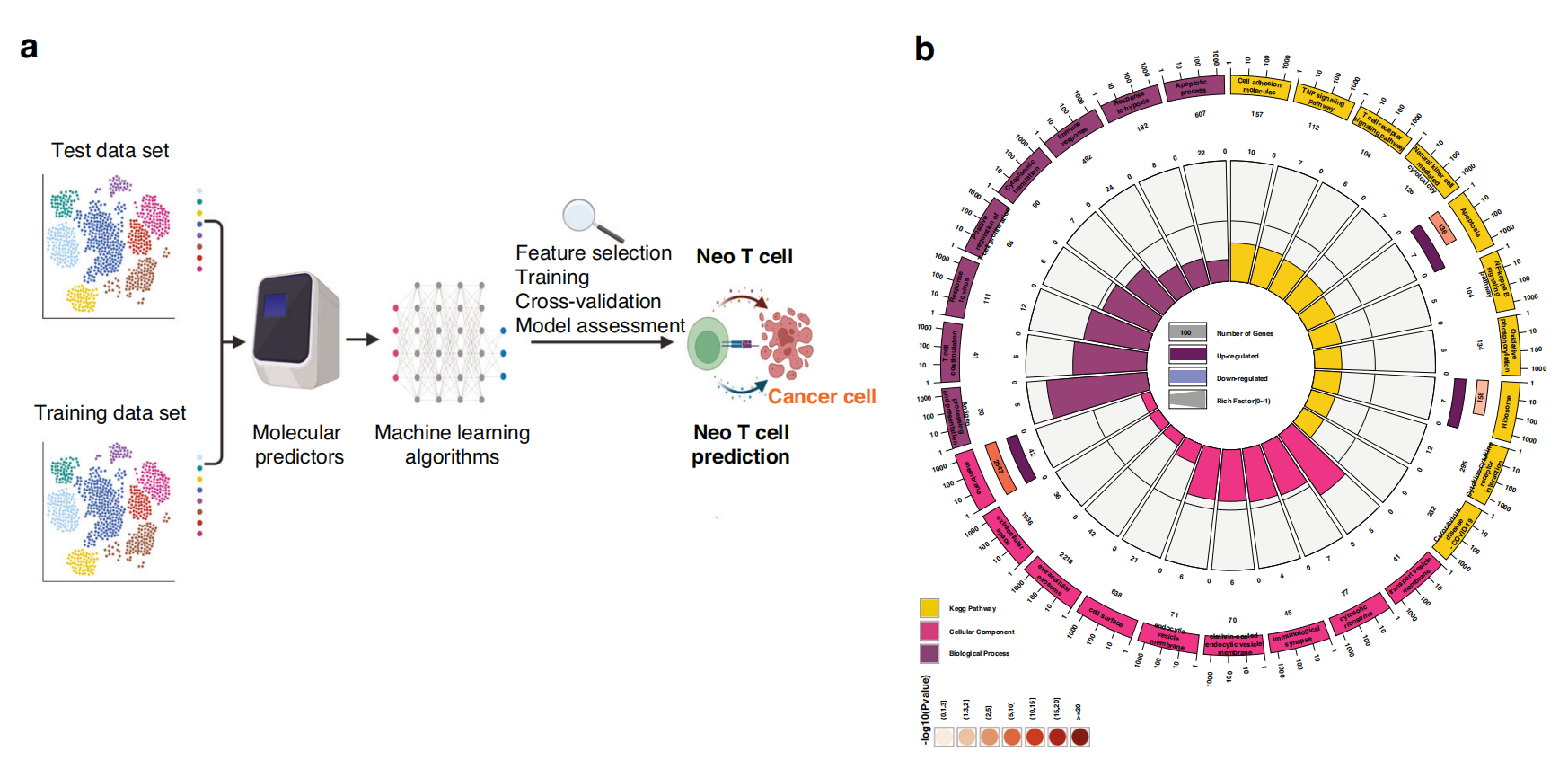


以下是对图中各个部分的分析:
a. 机器学习数据处理流程:这部分描述了用于处理单细胞测序数据的机器学习模型的数据流。这包括数据的预处理、标准化、特征选择等步骤,以准备数据进行模型训练。
b. KEGG和基因本体(Gene Ontology, GO)分析:对初步XGBoost模型中重要性指数大于0的168个特征基因进行了分析。外部的第二个环表示信号通路中的基因数量,内环中的柱状图高度表示特征基因在信号通路中总基因数的比例。这表明了这些基因在不同信号通路中的分布和重要性。
c. 特征基因的直方图:展示了26个重要性指数大于0.01的特征基因。这些基因被认为是Neo T细胞的关键标记基因。
d. 26个特征基因的网络表示:构建了一个蛋白质-蛋白质相互作用(PPI)网络,展示了这26个基因之间的关系。图中用红色突出显示的基因表示具有高特征重要性指数并且与其他分子频繁相互作用的基因。
e. Spearman相关性分析:分析了26个特征基因与它们所涉及的信号通路之间的Spearman相关性,颜色梯度表示Spearman相关系数的大小。这有助于理解这些基因如何与细胞信号传导途径相互作用,以及它们在Neo T细胞中的潜在功能。
总体而言,图1提供了一个综合的视角,展示了通过机器学习模型识别出的与Neo T细胞相关的基因,并分析了这些基因在生物学功能和信号通路中的作用。通过这种分析,研究人员可以更好地理解Neo T细胞的特性,并发现新的药物靶点或生物标志物。
五、机器学习+单细胞RNA测序,进行泛癌种的免疫治疗与生存预后分析

文献概述
这篇文章是一项关于癌症免疫治疗的研究成果,发表在《Journal of Experimental & Clinical Cancer Research》。
研究背景与目的:
免疫治疗已成为一种有效的癌症治疗手段,但只有部分患者能够从中受益。研究者们发现,肿瘤细胞中的乳酸代谢(Lactate Metabolism, LM)与免疫治疗的响应性有重要关联,因此探索通过靶向乳酸代谢来增强抗肿瘤免疫反应作为一种有前景的治疗策略。
研究方法:
- 收集并分析了接受免疫治疗的患者的公共单细胞RNA测序(scRNA-seq)队列,以确定乳酸代谢与免疫治疗反应之间的关系。
- 通过40个泛癌种scRNA-seq队列的广泛检查,制定了一个新的与乳酸代谢相关的特征签名(LM.SIG)。
- 使用多种机器学习(ML)算法,基于8个免疫治疗转录组队列和30个癌症基因组图谱(The Cancer Genome Atlas, TCGA)泛癌种数据集,验证了LM.SIG在预测免疫治疗反应和生存预后方面的能力。
- 基于17个CRISPR数据集,鉴定了潜在的免疫治疗靶点,并通过体内外实验进行了验证。
研究结果:
- 乳酸代谢的评估在两个免疫治疗scRNA-seq队列中证实与免疫治疗抗性有显著关系。
- 泛癌种数据大规模分析显示,LM.SIG与抗肿瘤免疫及免疫细胞的不平衡浸润呈负相关,而与促肿瘤信号呈正相关。
- 使用该特征签名,机器学习模型在验证集和测试集中预测免疫治疗反应和预后的AUC值分别达到0.73/0.80和0.70/0.87。
- LM.SIG在多种癌症中的预测性能优于已发布的签名。
- 通过CRISPR筛选,LDHA被识别为泛癌种生物标志物,用于评估免疫治疗反应和生存概率,并通过免疫组化(IHC)和空间转录组学(ST)数据集进一步验证。
- 实验表明,胰腺癌中LDHA的缺乏增强了CD8+ T细胞的抗肿瘤免疫反应,并改善了巨噬细胞的抗肿瘤极化,从而增强了免疫治疗的效果。
结论:
研究揭示了乳酸代谢与免疫治疗抗性的紧密联系,并进一步建立了泛癌种的LM.SIG,这成为选择适合免疫治疗患者的有力工具。
研究限制:
文章也提到了一些研究限制,例如在GSE115978数据集中只有治疗前和无反应的患者,以及需要通过更多实验来进一步验证LM.SIG和LDHA在泛癌种水平上的预测性能。
重点关注
研究的主要步骤和方法。
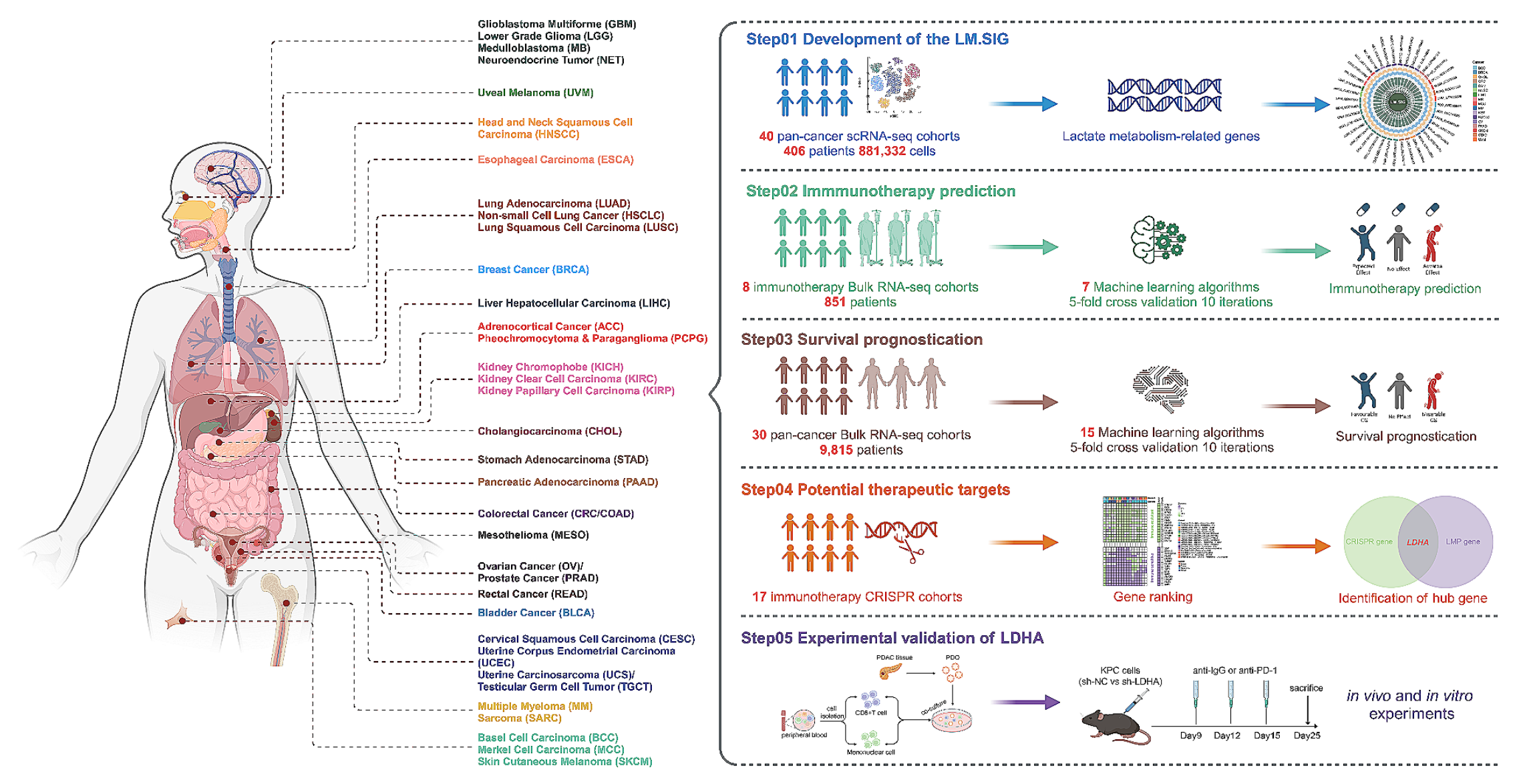
以下是对流程图的分析:
-
起始点:研究从收集和筛选单细胞RNA测序(scRNA-seq)数据集开始,这些数据集来自接受免疫治疗的患者。
-
数据整合:将筛选出的scRNA-seq数据集进行整合,以探索乳酸代谢(LM)与免疫治疗反应之间的关联。
-
特征签名开发:基于40个泛癌种scRNA-seq数据集,开发了一个与乳酸代谢相关的特征签名(LM.SIG)。
-
机器学习验证:使用多种机器学习(ML)算法对LM.SIG的预测能力进行验证,这些算法应用于8个免疫治疗转录组队列。
-
生存预后模型:利用SurvBenchmark设计,评估了15种不同的机器学习算法,以确定LM.SIG在生存预后方面的表现。
-
CRISPR筛选:使用17个CRISPR数据集来识别可能的免疫治疗靶点。
-
实验验证:通过体外和体内实验来验证潜在靶点的功能,特别是LDHA基因在胰腺癌中的作用。
-
结果分析:对所有步骤中得到的数据进行分析,以确定LM.SIG与免疫治疗反应和生存预后之间的关系。
-
结论:基于分析结果,得出乳酸代谢与免疫治疗反应之间存在显著关联的结论,并提出LM.SIG作为预测工具的潜力。
-
科学交流:研究结果通过论文发表,与科学界共享发现和方法。
整个流程图体现了从数据收集到实验验证,再到结果分析和科学交流的完整研究过程。通过这一流程,研究团队能够系统地评估乳酸代谢在癌症免疫治疗中的作用,并探索其作为预测和治疗靶点的潜力。
六、教师-学生协作的多实例学习框架,从12类肿瘤病理切片中预测PDL1的表达

文献概述
这篇文章描述了一种新的弱监督学习方法,名为MILTS(教师-学生协作的多实例学习框架),用于从常规的H&E染色组织切片中预测PDL1(程序性死亡配体1)的表达。
PDL1是一个重要的生物标志物,通常通过免疫组化(IHC)进行量化,但缺乏已建立的组织病理学模式。深度学习有助于组织病理学评估,但组织异质性和缺乏空间解析注释给精确分析带来了挑战。
MILTS利用批量RNA测序数据,通过教师-学生框架扩展了多实例学习范式,动态分配伪标签以解决切片内异质性,并使用时间集成模型蒸馏检索未标记实例。该方法在12,299张切片上进行了评估,涵盖了20种实体瘤类型,对于9种已建立PDL1作为生物标志物的肿瘤,在新鲜冷冻和甲醛固定标本上的加权平均曲线下面积(AUC)分别为0.83和0.74。MILTS预测的PDL1表达模式在20张切片上通过IHC验证,提供了与PDL1相关的组织学见解。这表明深度学习在从H&E图像中识别分子变化的多样化组织学模式方面具有潜力。
文章还讨论了PD1-PDL1检查点抑制剂在过去十年中彻底改变了癌症治疗,目前有7种抗PD1/PDL1单克隆抗体(mAbs)已获得美国食品药品监督管理局(FDA)的批准,还有大约六千种mAbs正在进行临床试验。PDL1的表达作为抗PD1/PDL1 mAbs(如pembrolizumab和nivolumab)治疗反应的生物标志物,是确定适合PD1-PDL1免疫治疗的患者队列的最广泛采用的标准。
研究结果表明,MILTS在预测PDL1表达方面优于其他方法,平均AUC提高了9%以上。通过对结直肠癌(COAD)、皮肤黑色素瘤(SKCM)和胃癌(STAD)数据的消融研究,发现教师-学生MIL模块和特征聚合模块对模型性能有显著提升。此外,MILTS在CPTAC数据集上进行了外部验证,尽管所有算法在应用于CPTAC数据集时性能有所下降,但MILTS仍然显示出比其他方法更优越的整体性能。
文章还探讨了PDL1表达的异质性模式,以及深度形态学特征如何帮助发现PDL1表达的形态类型。
例如,在结直肠癌中,高PDL1表达的区域通常与混合性炎症基质和相对丰富的嗜酸性粒细胞共存,并表现出肿瘤细胞的筛状生长模式。此外,还分析了MILTS预测与肿瘤免疫微环境和临床特征之间的相关性,发现M1型巨噬细胞和CD8+细胞毒性T细胞等细胞类型的存在与预测的PDL1表达呈正相关。
最后,文章讨论了MILTS在其他11种肿瘤类型中的性能,这些肿瘤类型尚未批准PDL1伴随测试或PDL1表达的预后意义尚未得到显著验证。研究结果表明,即使在PDL1表达水平较低的情况下,某些肿瘤类型也显示出与PDL1表达良好的组织学预测能力,这意味着除了PDL1表达水平之外,还有其他因素影响预测能力。
文章的方法部分详细描述了数据集、数据预处理、教师-学生协作的多实例学习(MILTS)方法、以及如何通过融合切片标记和切片级别预测的统计摘要来增强患者级别诊断。实现细节包括数据预处理、模型训练和评估的平台和编程环境。最后,文章提供了数据和代码的可用性信息。
重点关注
图1展示了MILTS(教师-学生协作的多实例学习框架)的工作流程以及其在临床上与PDL1相关的肿瘤上的性能表现。

该图分为三个部分:
a. MILTS的训练和推理工作流程包括三个主要步骤:
- 首先,将患者队列的数据分为训练集、验证集和测试集。
- 接着,对数据进行分块(patching)和随机增强(random augmentations)处理。
- 然后,使用得到的图像块(tile)来训练一个以多实例学习(MIL)方式工作的教师-学生协作网络。训练完成后,教师模型或学生模型将作为统计特征和深度特征的提取器。同一切片中的图像块的深度特征进一步融合成切片标记(slide token),并与图像块级别特征的统计摘要结合起来,通过训练一个多层感知器(MLP)分类器来进行患者级别的诊断。
图中还解释了MIL中的一些术语,例如:
- S 代表学生(student)
- T 代表教师(teacher)
- C 代表连接(concatenation)
- MLP 代表多层感知器(multi-layer perceptron)
b. 展示了不同肿瘤类型的切片图像数量。
c. 通过图表展示了模型在FFPE(甲醛固定石蜡包埋)切片和新鲜冷冻(fresh-frozen)切片上的性能,分别针对前述肿瘤类型。性能指标可能包括曲线下面积(AUC)、准确率、敏感度和特异性等。
从这个框架中,我们可以理解MILTS模型的创新之处在于其结合了教师-学生协作学习机制和多实例学习框架,以及如何利用深度学习来提取和融合特征,最终实现对PDL1表达水平的准确预测。这种预测对于癌症治疗和免疫疗法的决策具有重要意义。
















 1083
1083











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









