







小罗碎碎念
在医学AI领域,急性髓系白血病(AML)的基因检测意义重大。
目前,AML的精准风险分层依赖基因检测,像NPM1和FLT3-ITD突变的检测至关重要,但传统检测方法受资源限制,而基于全切片图像(WSI)的深度学习虽有潜力,却面临图像复杂和标注困难等问题。
为突破困境,研究团队构建了基于多实例学习(MIL)和集成技术的深度学习模型。

他们从572例患者获取WSI和基因数据,通过独特的数据处理流程,如三步细胞图像生成、数据上采样平衡数据等,训练模型。

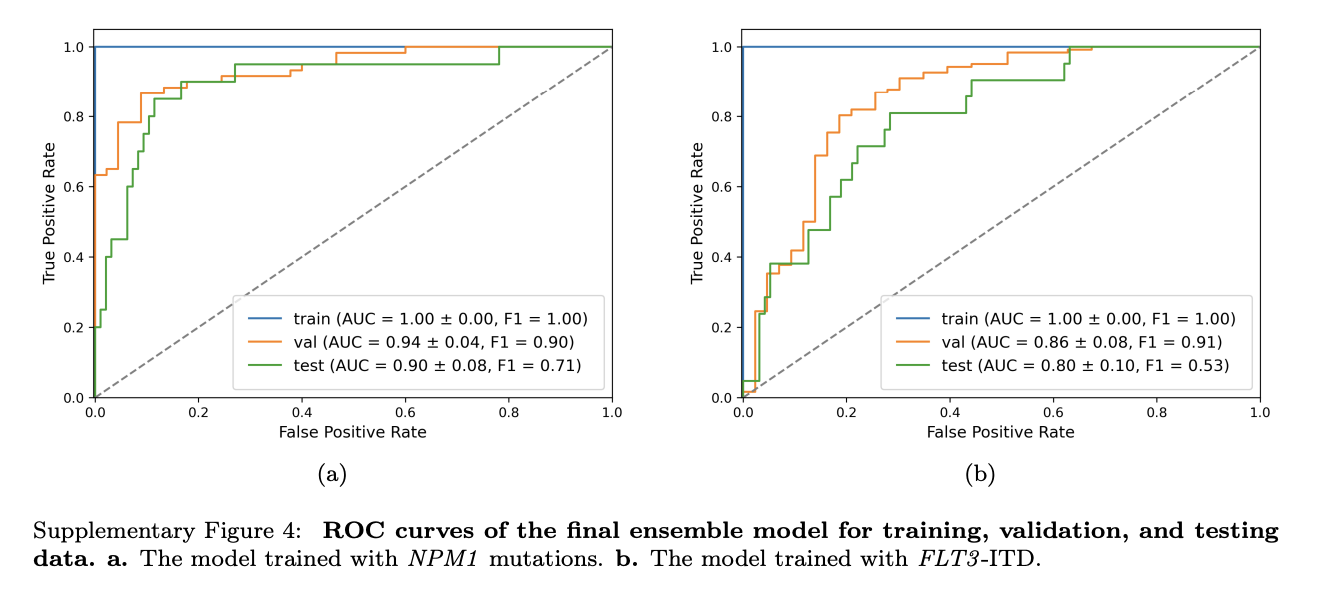
结果显示,该模型在预测NPM1和FLT3-ITD突变上表现出色,AUC分别达到0.90 ± 0.08和0.80 ± 0.10,且发现blasts细胞对突变预测意义关键。
这一研究成果为医学AI研究提供了新思路。其方法在无需细胞级手动标注下实现高精度预测,节省人力。
不过,模型仍有提升空间,如降低FLT3-ITD突变的假阳性率。后续研究可从嵌入更多基因特征、提升图像分辨率着手,这将推动医学AI在白血病诊断领域进一步发展,助力实现更精准的临床诊断和治疗。
交流群
欢迎大家加入【医学AI】交流群,本群设立的初衷是提供交流平台,方便大家后续课题合作。
目前小罗全平台关注量61,000+,交流群总成员1400+,大部分来自国内外顶尖院校/医院,期待您的加入!!
由于近期入群推销人员较多,已开启入群验证,扫码添加我的联系方式,备注姓名-单位-科室/专业,即可邀您入群。
知识星球
对推文中的内容感兴趣,想深入探讨?在处理项目时遇到了问题,无人商量?加入小罗的知识星球,寻找科研道路上的伙伴吧!
一、文献概述
文章提出一种基于多实例学习(MIL)和集成技术的深度学习模型,可从急性髓系白血病(AML)的全切片图像(WSI)预测基因突变,为临床诊断提供支持。
1-1:研究背景
AML是一种侵袭性血液恶性肿瘤,NPM1和FLT3-ITD突变常见且对治疗意义重大,但现有分子检测存在资源不均和耗时久的问题。
深度学习虽在医学图像处理有进展,但分析骨髓穿刺液WSI面临细胞结构复杂、手动标注繁琐等挑战。
1-2:研究方法
- 数据集:纳入572例AML患者的WSI和基因突变数据,经染色和扫描处理,确定基因突变状态。
- 细胞图像生成:对WSI骨髓涂片经三步处理获取细胞图像,依次是用PyHIST工具生成补丁、用基于DenseNet121的模型选择ROI补丁、用基于YOLOv4的模型检测白细胞。
- 数据增强:针对数据不平衡,将突变WSI细胞分成多组(袋),每组2000个细胞;标准WSI随机选2000个细胞为一袋,平衡数据集并加速训练。
- 多实例学习:MIL将二进制分类任务转化为基于“袋”的学习问题,训练分推理和学习阶段,用交叉熵损失函数更新模型权重。
- 集成学习:采用损失加权法,选取损失值最低的前三个MIL模型组合,增强模型预测能力。
1-3:研究结果
- ROI选择和白细胞检测:开发的管道能有效选择ROI补丁,减少后续处理数据量。YOLOv4模型检测白细胞,设置相关参数确保选到高质量细胞。
- 模型性能:细胞级上采样结合集成学习提升模型性能,NPM1突变和FLT3-ITD预测的AUC分别达0.90±0.08和0.80±0.10,且降低了假阳性率。
- 细胞特征重要性:blasts是预测基因突变的关键指标,突变样本中blasts比例高,非突变样本中比例低。
二、建模思路
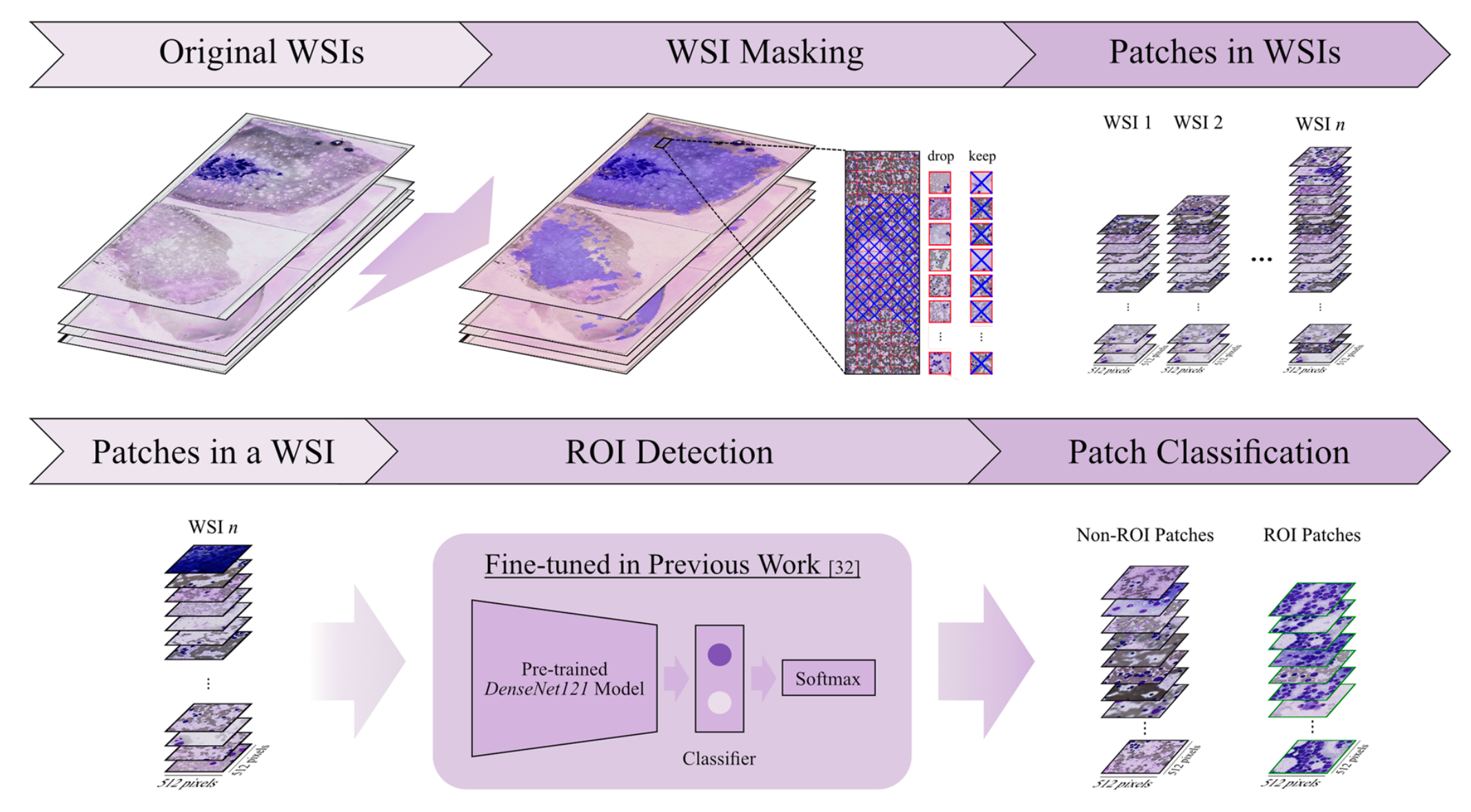
这是从急性髓系白血病骨髓涂片全切片图像(WSI)中处理图像块(Patches)的部分流程,主要步骤如下:
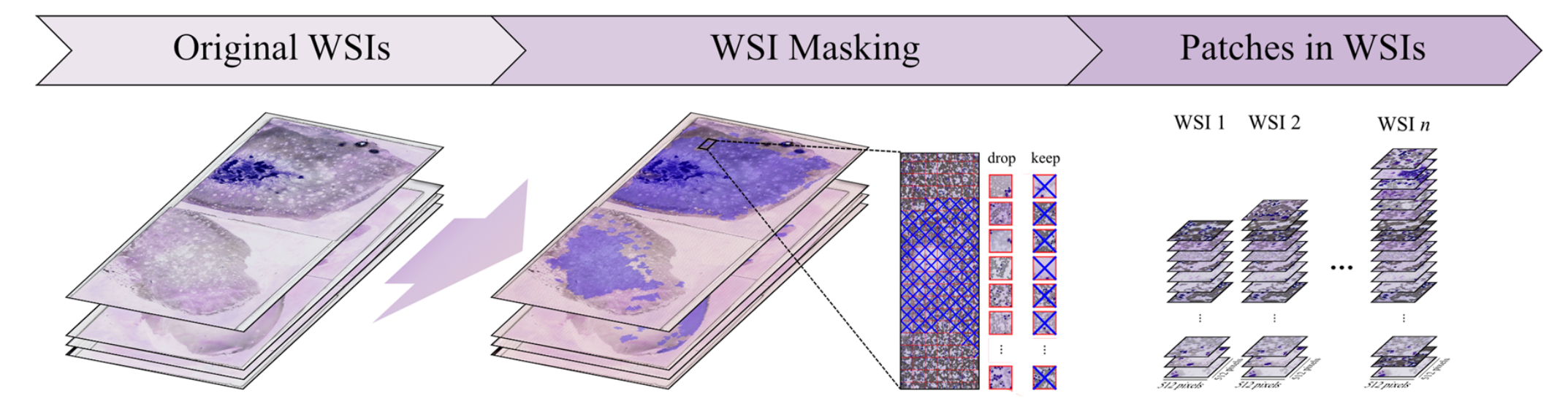
2-1:WSI掩膜处理
原始全切片图像(Original WSIs)经过WSI掩膜操作,生成一系列图像块(Patches in WSIs) 。
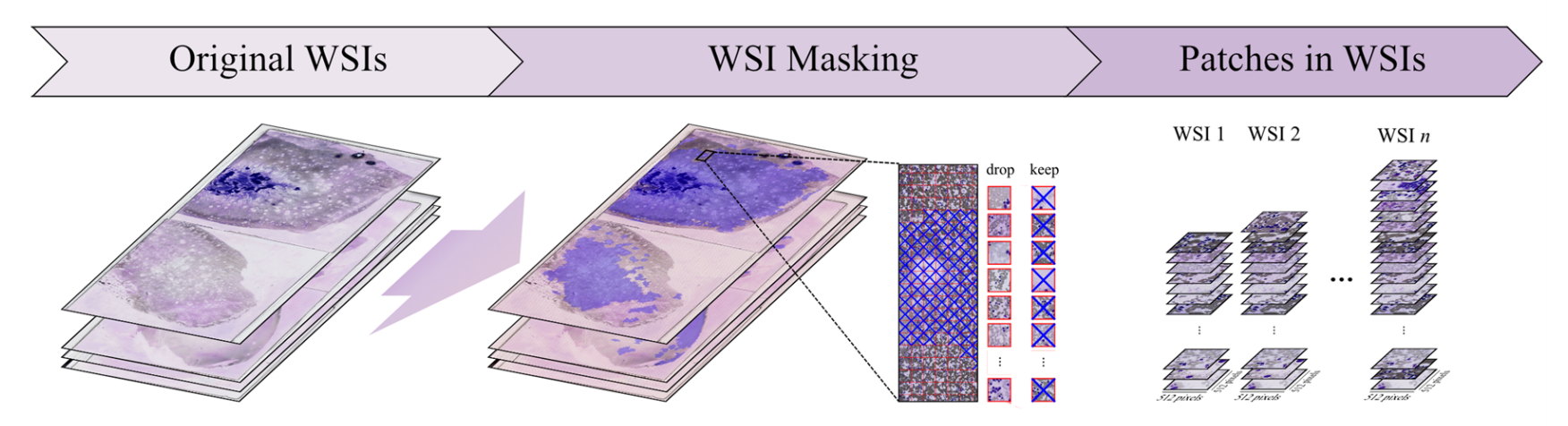
在这个过程中,对生成的图像块进行筛选,决定保留(keep)或丢弃(drop)某些图像块,以获取合适的图像块集合用于后续分析。
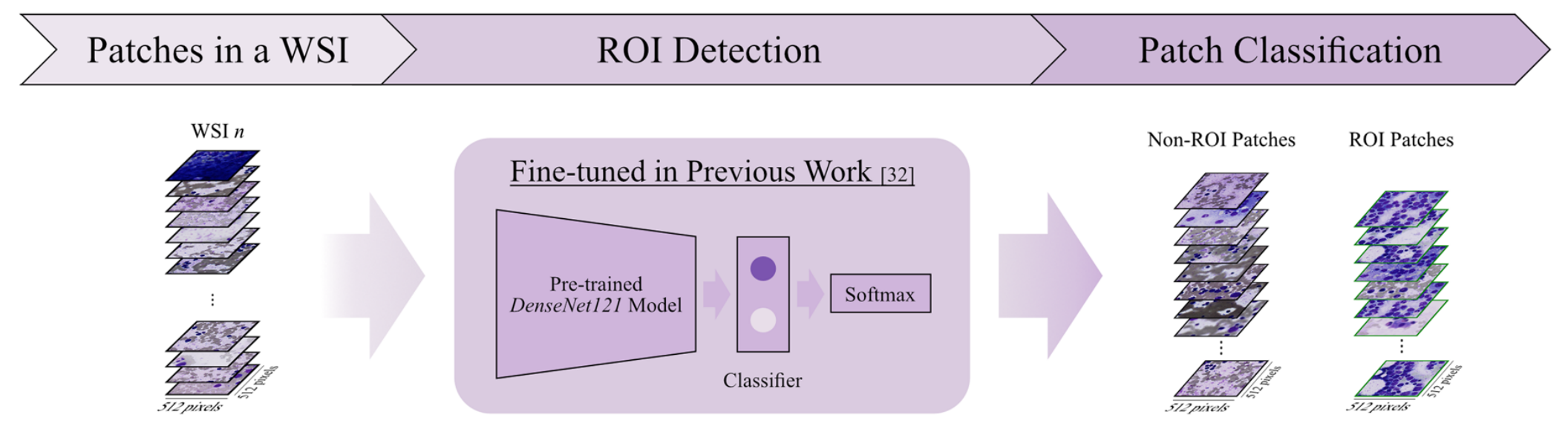
2-2:感兴趣区域检测
将一个WSI中的图像块(Patches in a WSI)输入到基于先前工作[32]微调过的预训练DenseNet121模型中。
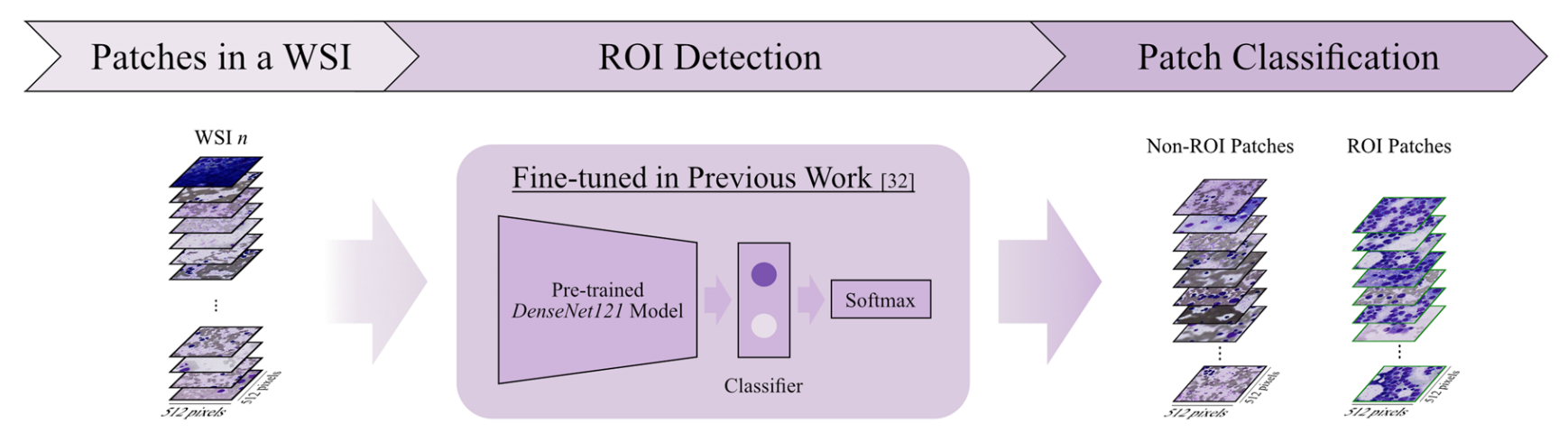
该模型结合Softmax分类器,对输入的图像块进行分类,从而区分出感兴趣区域(ROI)图像块和非ROI图像块 。
这一步有助于聚焦于对后续分析更有价值的图像区域,去除无关或干扰性的图像部分。
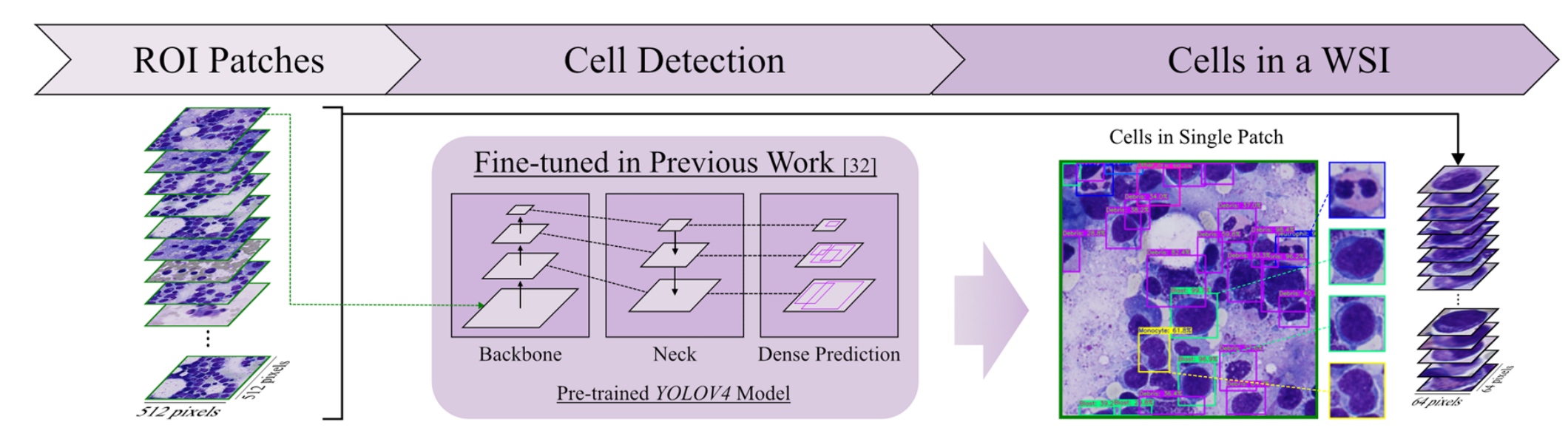
2-3:细胞检测
将之前得到的感兴趣区域图像块(ROI Patches)输入到基于先前工作[32]微调过的预训练YOLOv4模型。
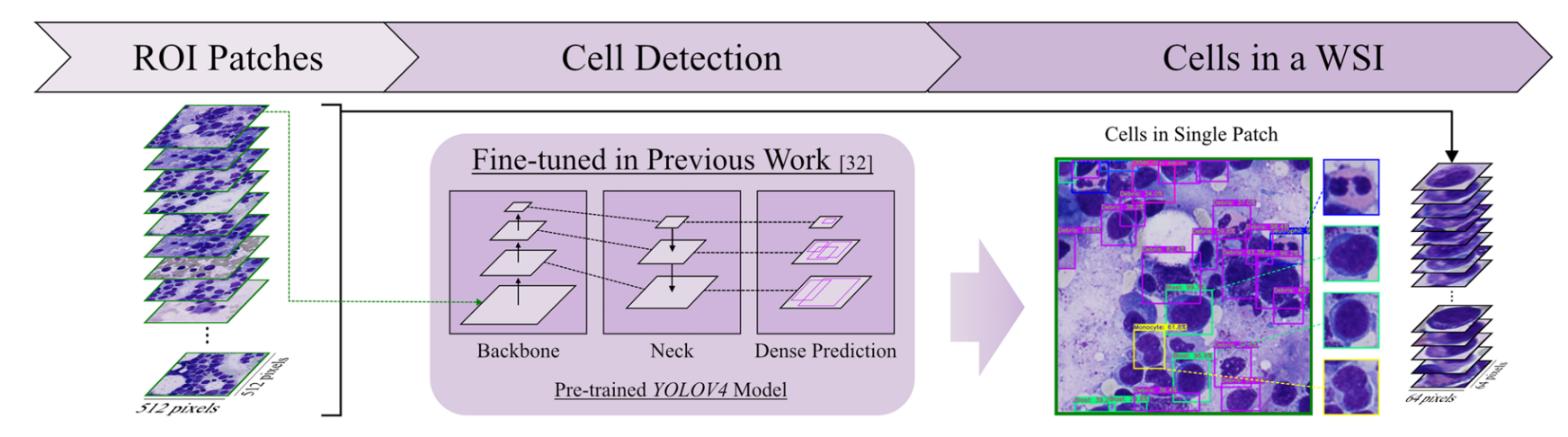
该模型由Backbone、Neck和Dense Prediction等部分构成,能检测出ROI Patches中的白细胞细胞,得到单个图像块中的细胞(Cells in Single Patch),进而获取全切片图像中的细胞集合(Cells in a WSI) 。
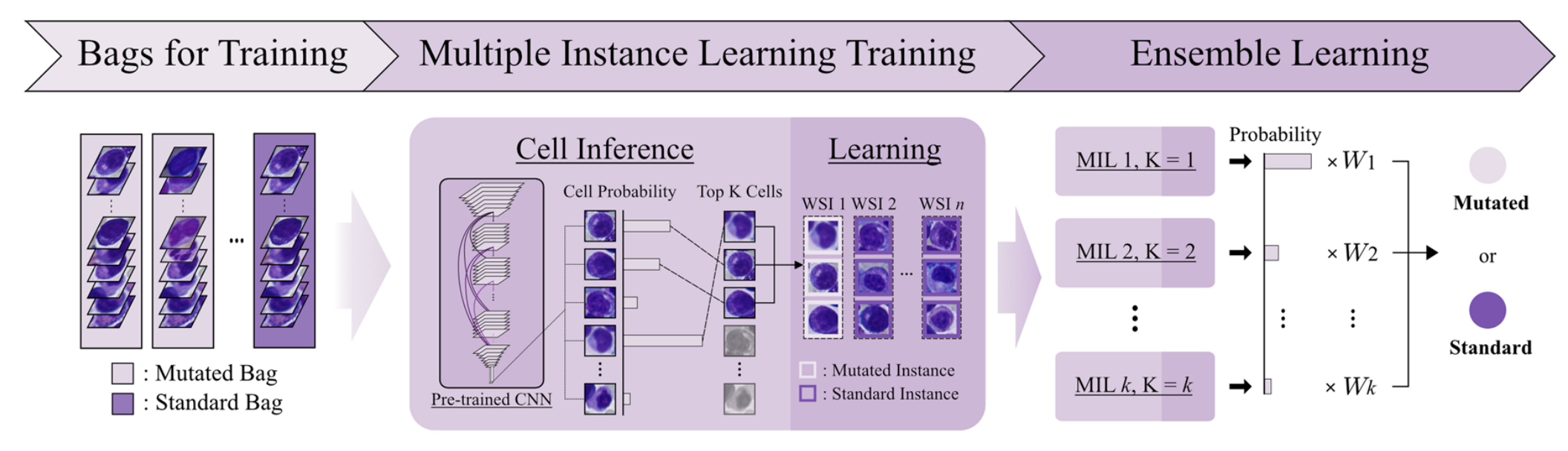
2-4:数据上采样及成袋处理
针对全切片图像中的细胞(Cells in WSIs) ,由于突变WSI(Mutated WSI)和标准WSI(Standard WSI)数量可能不均衡,为平衡数据,将突变WSI随机选取细胞组成多个“袋”(Bag),即一个突变WSI被划分成多个突变袋 。

同时,从标准WSI中也随机选取细胞组成袋。
这些袋(Bags for Training)将用于后续的多实例学习训练,袋分为带有突变标签(Mutated Label)的袋和带有标准标签(Standard Label)的袋 。
2-5:模型训练及预测
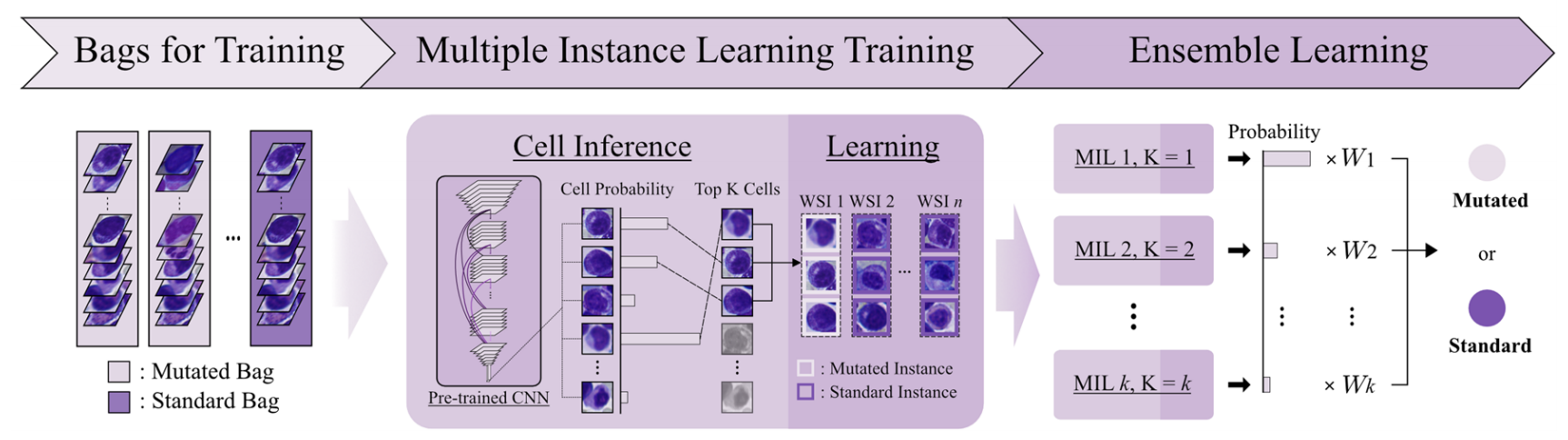
多实例学习训练
- 细胞推理(Cell Inference):将训练用的“袋”(Bags for Training ,含突变袋和标准袋)输入到预训练的CNN模型中。模型计算每个细胞的突变概率(Cell Probability),并依据概率对细胞进行排名,选取排名前K的细胞(Top K Cells) 。
- 学习(Learning):针对每个“袋”,基于选取出的前K个细胞,通过对数据集的全推理过程,依据细胞突变概率排名来学习这些细胞,进而更新模型权重 。
集成学习
训练得到多个不同参数设置(如MIL 1, K = 1;MIL 2, K = 2等 )的多实例学习(MIL)模型。
通过对这些模型预测结果按照一定权重(W1、W2、…、Wk )进行加权组合,综合各模型优势,最终输出样本是突变(Mutated)或标准(Standard)状态的预测结果 。
三、方法
3-1:数据集
1994年至2015年间,572例在国立台湾大学医院(NTUH)确诊为新发急性髓系白血病(AML)的患者被纳入本研究。
骨髓涂片和外周血涂片经改良Romanowsky染色后,使用配备40x NA 0.75物镜的滨松NanoZoomer数字玻片扫描仪扫描成全切片图像(WSIs),分辨率约为0.23 μm/像素。
在HiSeq平台(Illumina,加利福尼亚州圣地亚哥)上使用TruSight骨髓检测组确定基因突变状态 ,突变标注为1(表示存在致病性或可能致病性突变)或0(表示不存在此类突变) 。
在本队列中,共发现34个基因经常发生突变,本研究列出突变频率超过10%的基因,并选择突变频率最高的前两个作为研究目标(表1)。

3-2:细胞图像生成
WSIs的骨髓涂片需经过三步过滤过程来识别细胞,以供后续模型训练。
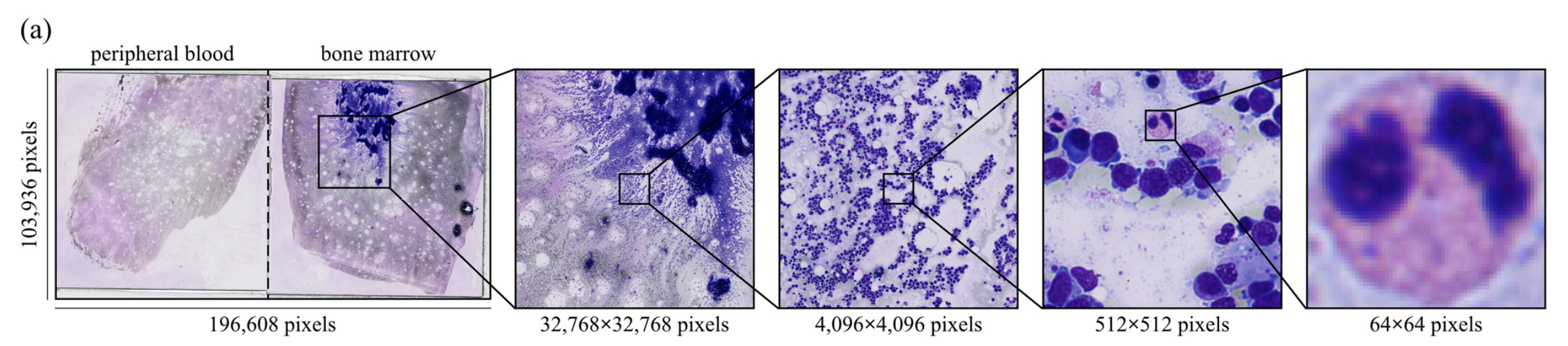
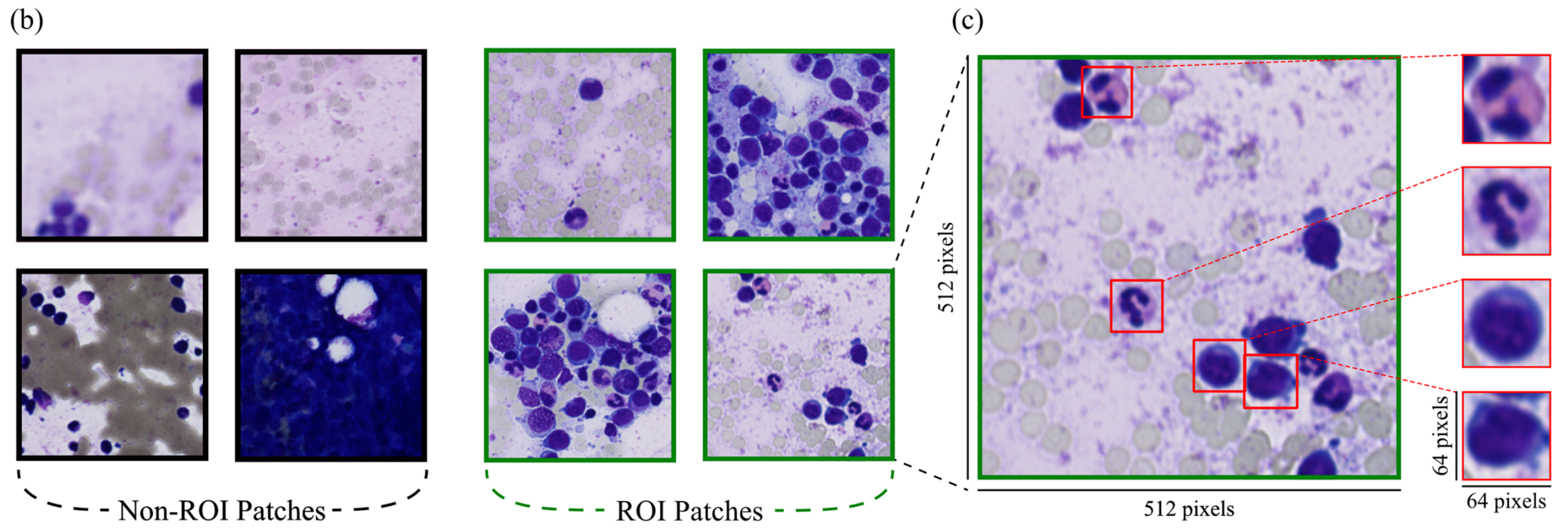
首先,使用PyHIST工具生成图像块 。该工具用于过滤掉WSIs中的背景区域和非涂片区域(图1a)。
以最高分辨率(40倍放大)提取512×512像素的图像块,在PyHIST中使用基于图的分割方法 作为“生成方法”参数,内容阈值设为0.05。
“tilecross-downsample”和“mask-downsample”参数采用默认值,“output-downsample”参数设为1,以获取原始分辨率的图像块。
其次,由PyHIST工具生成的图像块进一步分为感兴趣区域(ROI)和非ROI图像块。由于缺乏手动标注的ROI和非ROI数据,作者使用从先前研究获得训练权重的ROI检测模型 。该模型基于DenseNet121 ,在参考文献[31]中标记的图像块(ROI/非ROI)上进行预训练和微调。
在此步骤(图1b)中,去除含有密集白细胞或无白细胞的图像块,显著减少了后续细胞检测建模步骤的输入数据量。
在ROI选择之后,每个WSI中的ROI图像块将用于第三步获取细胞。作者应用从同一研究获得训练权重的细胞检测模型 。
该模型基于YOLOv4 ,在参考文献[31]中经过训练以预测白细胞目标的边界框。在此步骤中,检测骨髓穿刺液每个ROI图像块中的所有白细胞(图1c)。
3-3:数据上采样
WSI队列存在显著的类别不平衡,与大量标准样本相比,突变样本相对较少。
这种不平衡可能影响训练结果,因为模型可能会偏向于将所有WSIs预测为“标准”,从而忽略突变病例。
为解决这种不平衡,作者引入上采样策略,将突变WSI的细胞划分为多个集合(袋),每个集合包含固定数量的细胞(图1d)。

先前研究表明,500个细胞的集合足以捕捉骨髓细节 。在本研究中,作者将此数量增加四倍至2000个细胞,以增强每个WSI的代表性。
在多实例学习(MIL)训练期间,这些袋被视为独立样本,有效增加了训练数据中的突变类别,使数据集更加平衡。
除了解决数据不平衡问题,作者还面临标准WSIs的挑战,这些样本未进行上采样且每个包含数万个细胞,大量的输入数据会显著减慢模型训练速度。
为解决此问题,作者从每个标准WSI中随机选择固定数量(2000个)的细胞作为一个袋,通过减少输入量来加快训练过程。
阳性全切片图像(WSIs)至少包含一个阳性实例,而阴性WSIs不包含任何阳性实例。
在本研究中,“袋(bags)”指上采样后创建的袋,“实例(instances)”表示袋中的单个细胞。
经过上述细胞检测过程,生成袋 B = { B i ; i = 1 , 2 , … , n } B = \{B_i; i = 1, 2, \ldots, n\} B={Bi;i=1,2,…,n} ,其中 B i = { c i , 1 , c i , 2 , … , c i , m i } B_i = \{c_{i,1}, c_{i,2}, \ldots, c_{i,m_i}\} Bi={ci,1,ci,2,…,ci,mi} 是包含 m i m_i mi 个细胞的袋(图1e)。
大多数袋中 m i = 2000 m_i = 2000 mi=2000 ,但来自突变WSI的单个袋可能包含少于2000个细胞,因为WSI的细胞总数通常不是2000的精确倍数。
作者根据袋来源WSI的标签将袋标记为“突变”或“标准”。在一个袋中,使用嵌入模型对所有细胞进行分类,其训练过程在以下段落详细说明。然后根据细胞的突变概率对细胞进行排名。MIL任务涉及学习一种细胞级嵌入,以有效识别并区分突变袋中的特征细胞与标准细胞。
在训练过程中(图1e),MIL训练过程由两个交替阶段组成:推理阶段和学习阶段。
首先,采用DenseNet121模型并进行微调作为嵌入模型。它使用PyTorch库提供的预训练权重初始化,这些权重先前在ImageNet数据集上进行过训练。
该模型表示为函数 f θ f_\theta fθ ,当前参数 θ \theta θ 将输入细胞 c i , j c_{i,j} ci,j 映射到“突变”概率。在MIL训练的细胞推理阶段,为袋 B i B_i Bi 导出向量列表 P i = { p i , 1 , p i , 2 , … , p i , m i } P_i = \{p_{i,1}, p_{i,2}, \ldots, p_{i,m_i}\} Pi={pi,1,pi,2,…,pi,mi} ,表示细胞 c i , j c_{i,j} ci,j ( j = 1 , 2 , … , m i j = 1, 2, \ldots, m_i j=1,2,…,mi )属于“突变”类别的概率。
在此阶段,模型函数 f θ f_\theta fθ 的权重被冻结,确保在生成估计概率时参数保持固定。
在MIL训练的学习阶段,作者引入超参数 K K K ,从每个袋中选择概率 ( p i , j ) (p_{i,j}) (pi,j) 最高的前 K K K 个细胞作为嵌入模型的训练数据。这些选定细胞的真实标签 ( y i , j ) (y_{i,j}) (yi,j) 根据其所在袋的标签确定:每个突变袋中的前 K K K 个细胞标记为突变细胞,标准袋中的前 K K K 个细胞标记为标准细胞。
然后,使用交叉熵损失函数
l
l
l (如公式(1)所示)将网络输出
y
~
i
,
j
=
f
θ
(
c
i
,
j
)
=
p
i
,
j
\widetilde{y}_{i,j} = f_\theta(c_{i,j}) = p_{i,j}
y
i,j=fθ(ci,j)=pi,j 与标签
y
i
,
j
y_{i,j}
yi,j 进行比较:
l
=
∑
y
~
i
,
j
−
w
1
[
y
i
,
j
log
(
y
~
i
,
j
)
]
−
w
0
[
(
1
−
y
i
,
j
)
log
(
1
−
y
~
i
,
j
)
]
l = \sum_{\widetilde{y}_{i,j}} -w_1[y_{i,j} \log(\widetilde{y}_{i,j})] - w_0[(1 - y_{i,j}) \log(1 - \widetilde{y}_{i,j})]
l=y
i,j∑−w1[yi,jlog(y
i,j)]−w0[(1−yi,j)log(1−y
i,j)]
其中
w
1
w_1
w1 是突变袋数量除以总袋数量,
w
0
=
1
−
w
1
w_0 = 1 - w_1
w0=1−w1 。
因此,基于损失函数公式(1) ,更新 f θ f_\theta fθ 的权重,以最小化预测输出与实际标签之间的差异。
3-4:集成学习
集成方法是一种强大的手段,涉及训练并组合多个模型以解决复杂问题。
集成方法的核心思想是多个单个的“弱学习器”协同工作,形成一个“强学习器” 。每个模型通过投票贡献自己的判断,集成方法则将这些输入结合起来生成最终预测。
集成的总体目标是通过利用多个模型的综合能力,而非仅依赖单个模型,来减少预测中的偏差和方差。
在本研究中,作者采用了一种基于损失的加权集成方法。在这种方法里,集成中每个模型的贡献由其单个损失函数 l M K l_{M_K} lMK(如公式(1)所示)决定 。
这种加权策略赋予损失值更低的模型更大的影响力,从而增强集成中更准确模型对最终概率 ( P e n s e m b l e ) (P_{ensemble}) (Pensemble)的影响 。仅选取根据损失值确定的前三个多实例学习(MIL)模型作为集成中的弱学习器。
此方法旨在优化基础模型的组合 { M K 1 , M K 2 , M K 3 } = a r g m i n K ∈ { M 1 , … , M 30 } \{M_{K_1}, M_{K_2}, M_{K_3}\} = argmin_{K \in \{M_1, \ldots, M_{30}\}} {MK1,MK2,MK3}=argminK∈{M1,…,M30},其中 K 1 K_1 K1、 K 2 K_2 K2和 K 3 K_3 K3对应于 { M 1 , … , M 30 } \{M_1, \ldots, M_{30}\} {M1,…,M30}中损失值最低的三个MIL模型 。
集成中的每个基础模型被赋予一个权重
W
i
W_i
Wi,由其性能按公式(2)确定:
W
i
=
e
x
p
(
−
l
i
)
∑
i
∈
{
M
K
1
,
M
K
2
,
M
K
3
}
e
x
p
(
−
l
i
)
W_i = \frac{exp(-l_i)}{\sum_{i \in \{M_{K_1}, M_{K_2}, M_{K_3}\}} exp(-l_i)}
Wi=∑i∈{MK1,MK2,MK3}exp(−li)exp(−li)
集成模型根据这三个模型各自的权重
(
W
M
K
)
(W_{M_K})
(WMK),将它们的概率
(
m
a
x
P
i
)
(max P_i)
(maxPi)进行组合,如公式(3)所示:
P
e
n
s
e
m
b
l
e
=
∑
i
∈
{
M
K
1
,
M
K
2
,
M
K
3
}
W
i
×
m
a
x
P
i
P_{ensemble} = \sum_{i \in \{M_{K_1}, M_{K_2}, M_{K_3}\}} W_i \times max P_i
Pensemble=i∈{MK1,MK2,MK3}∑Wi×maxPi
四、结果
4-1:感兴趣区域(ROI)图像块的自动选择
在数字病理学中,骨髓穿刺涂片的玻片通过数字玻片扫描仪扫描,生成高分辨率的全切片图像(WSIs),供血液病理学家分析(图2a)。
为启动该过程,从国立台湾大学医院获取的572例骨髓穿刺WSIs中采样数据集。为解决检测ROI图像块的问题,作者开发了一个分阶段选择ROI图像块的流程,自动识别骨髓穿刺WSIs中适合细胞学分析的区域。
单个WSI可能仅包含少数适合细胞学分析的区域。这些区域分布稀疏,细胞重叠和染色伪影极少,且具有细胞分类所需的细微复杂细胞学特征。
为高效获取此类区域,首先使用PyHIST工具 ,采用默认图形方法去除空白背景区域,仅保留染色部分。随后,作者采用微调的DenseNet121架构,利用先前研究的预训练权重 将单个图像块分类为ROI图像块或非ROI图像块(图2b)。
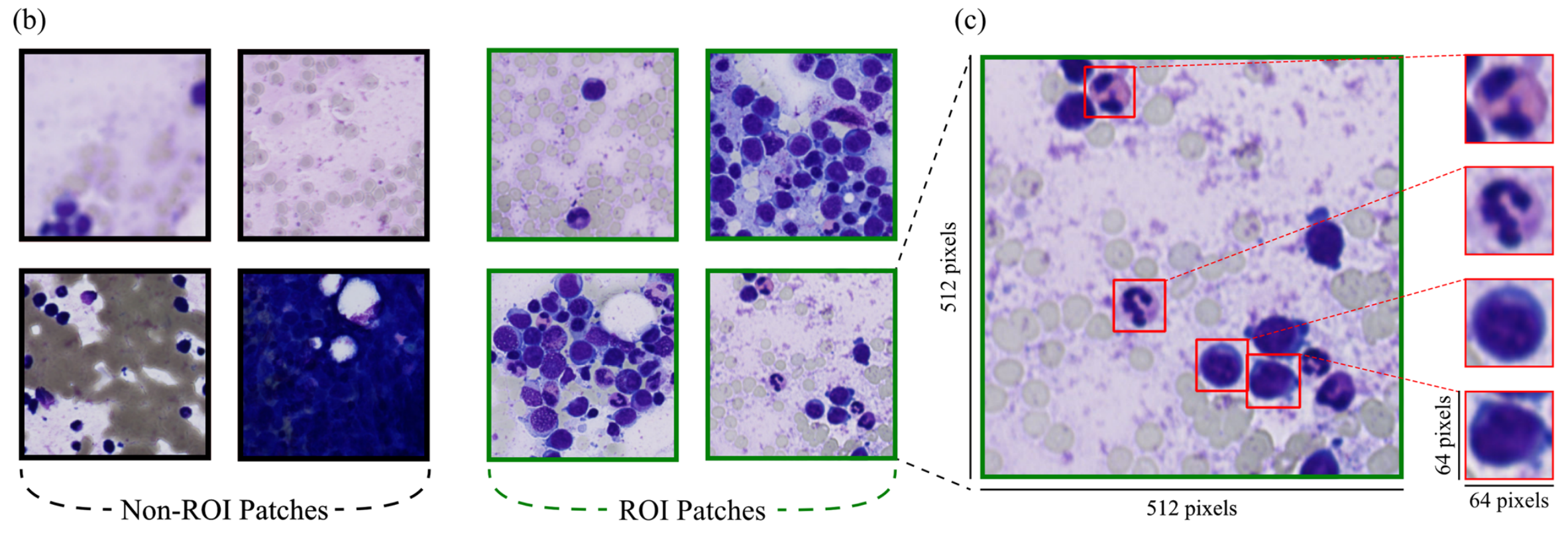
作者观察到分类结果符合预期,即在实际情况中,通常WSI中只有10 - 20%的区域可能是细胞学分析的ROI区域。
应用DenseNet121后的结果显示,对于大多数WSIs,经过此选择过程后,ROI图像块数量减少到原始数量的10 - 25%左右,表明图像块数量大幅减少(补充图1)。
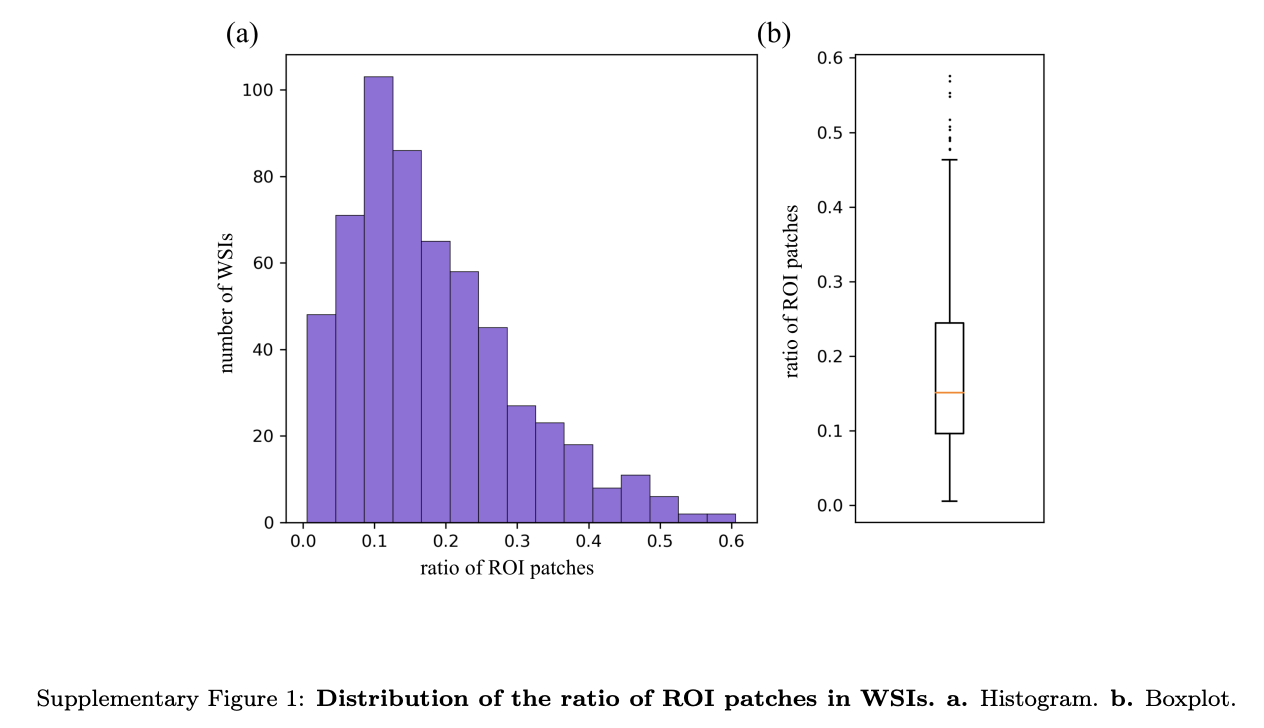
此筛选过程显著减少了后续细胞检测过程所需的时间,在去除指定范围内细胞过度重叠、组织伪影过多或无血细胞的区域方面,展现出出色的过滤效果。
这些有问题的图像块被有效去除,同时如补充图2所示,细胞数量少的图像块未被错误丢弃。
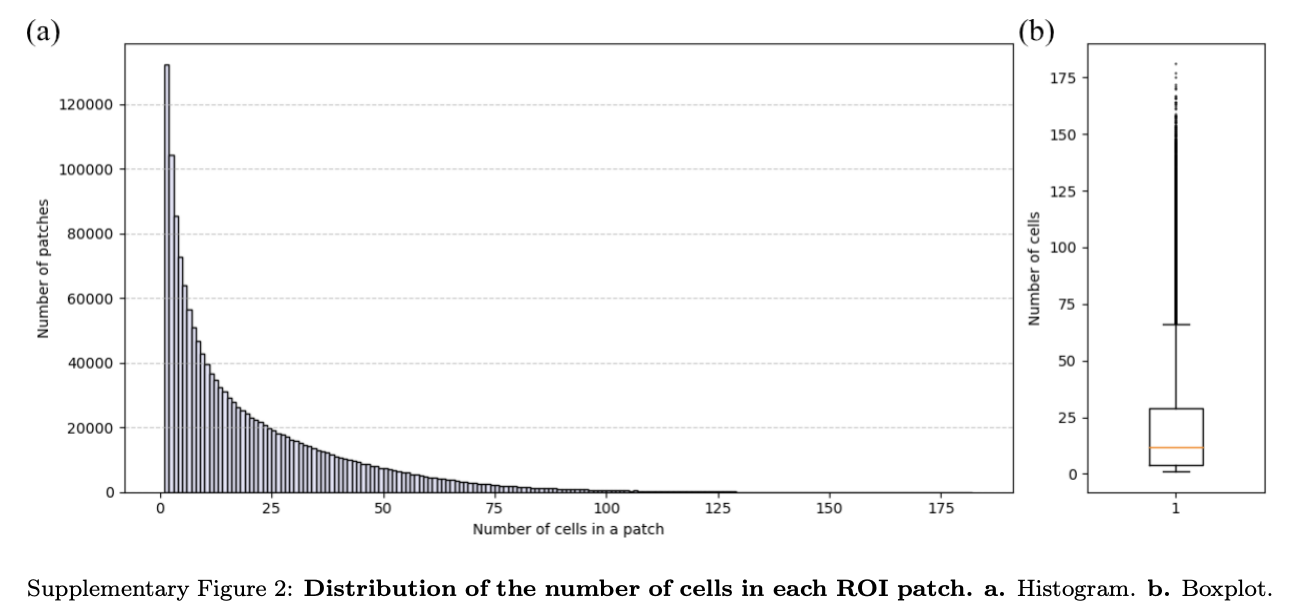
这种方法确保保留细胞数量少的图像块,支持数据完整性,避免不必要地丢失有价值信息(图2b)。
4-2:白细胞检测
在ROI选择过程之后,作者采用YOLOv4模型自动检测和分类所选ROI图像块中的细胞和非细胞物体。
在缺乏手动标注细胞的情况下,作者使用在同一研究中针对选择ROI图像块已预训练和微调过的训练权重 ,直接应用于该检测模型。
以ROI检测模型识别的ROI图像块作为输入,此YOLOv4模型旨在自动检测和分类骨髓样本中的所有细胞和非细胞物体。
作者进一步设置置信度阈值为0.5。置信度得分低于此阈值的细胞不被选取,确保为后续MIL模型训练选择高质量细胞。除管理置信度水平外,作者在过程中考虑了细胞大小。
在波长为400 nm的紫外光下,理论分辨率约为325.33 nm。作者将细胞大小范围设置为51×51像素到80×80像素。这意味着51像素的细胞大小约为16.6 μm,而80像素对应约25.6 μm。此范围是根据白细胞(WBCs)的典型大小(约10 - 25 μm)以及原始细胞(blasts,通常约15 - 20 μm)选择的。
通过将选择阈值设置在51到80像素之间,作者旨在捕获最具代表性、完整且与特征提取相关的细胞。所选细胞直接调整为64×64像素,以标准化输入大小用于MIL训练,避免其他填充方法可能引入的如相邻细胞或过多背景等无关特征(图2c)。
每个WSI剩余的细胞数量在100到100,000之间变化,大多数细胞数量低于20,000,平均为11,273(图3)。
这一结果在保持所有输入细胞最佳质量的同时,有效减少了MIL模型的输入量,加快了训练时间且不影响准确性。
因此,作者将包括嗜碱性粒细胞、原始细胞、嗜酸性粒细胞、淋巴细胞、晚幼粒细胞、单核细胞、中幼粒细胞、中性粒细胞和早幼粒细胞在内的白细胞作为MIL训练的输入,称为“所有细胞”。
在这些细胞中,MIL随机选择2000个细胞作为一个WSI的代表性细胞,即一个袋。在选择期间,作者对突变WSIs应用上采样技术以平衡数据。通过将每个突变WSI分离成多个袋,作者可以生成许多包含2000个细胞的突变袋。
4-3:细胞级多实例学习
将572例WSIs的总数据集在玻片层面按约7:1:2(400:56:116个WSIs)的比例划分为训练集、验证集和测试集。
作者使用DenseNet121作为基础嵌入模型,用PyTorch库基于ImageNet数据集训练提供的预训练权重进行初始化 。
MIL模型训练100个轮次,学习率设为0.0001。通过使用Adam优化器的随机梯度下降(SGD)实现损失最小化。
批量大小由每个袋内的细胞数量决定,限制为每批最多2000个实例(细胞)。由于上采样后训练数据仍存在轻微类别不平衡,对于NPM1突变,作者将权重 ( w 0 , w 1 ) (w_0, w_1) (w0,w1)设为 ( 0.51 , 0.29 ) (0.51, 0.29) (0.51,0.29) ,对于FLT3 - ITD设为 ( 0.37 , 0.63 ) (0.37, 0.63) (0.37,0.63) 。
为模拟真实场景,验证集和测试集不进行上采样。
每个轮次使用包含56个WSIs的不平衡验证集进行评估,即标准WSIs多于突变WSIs 。
采用提前终止策略防止过拟合。单独的测试集包括96个标准和20个NPM1突变WSIs ,以及95个标准和21个FLT3 - ITD WSIs。
除既定的MIL训练外,作者利用集成学习,在MIL中使用不同的 K K K值(从 K = 1 K = 1 K=1到 K = 30 K = 30 K=30 ),使模型对数据有不同视角。每个 K K K代表MIL训练过程中一个袋内考虑的正实例数量,从而创建对不同数据子集关注重点各异的模型。
基于公式(2)和公式(3) ,作者计算损失值最低的前3个MIL模型各自的权重 W i W_i Wi ,然后估计最终预测结果。模型在训练集、验证集和测试集上的预测性能见补充图4。
为阐明在MIL学习过程中细胞特征对预测的重要性,作者比较了九种细胞类型的比例表示。
在应用MIL模型前,每个WSI中的所有细胞类型数量除以该WSI中所有类型细胞的总数,以获得每种细胞类型的比例。
预测后,作者列出模型预测为与每个袋中突变相关性最高的概率前100的细胞图像。然后,根据细胞类型计算MIL预测的所有代表性细胞在原始WSI中的比例(图4)。
这张图(图4)展示了多实例学习(MIL)前后细胞类型比例的比较,通过箱线图呈现,分别针对NPM1突变数据(图a)和FLT3 - ITD数据(图b) 。
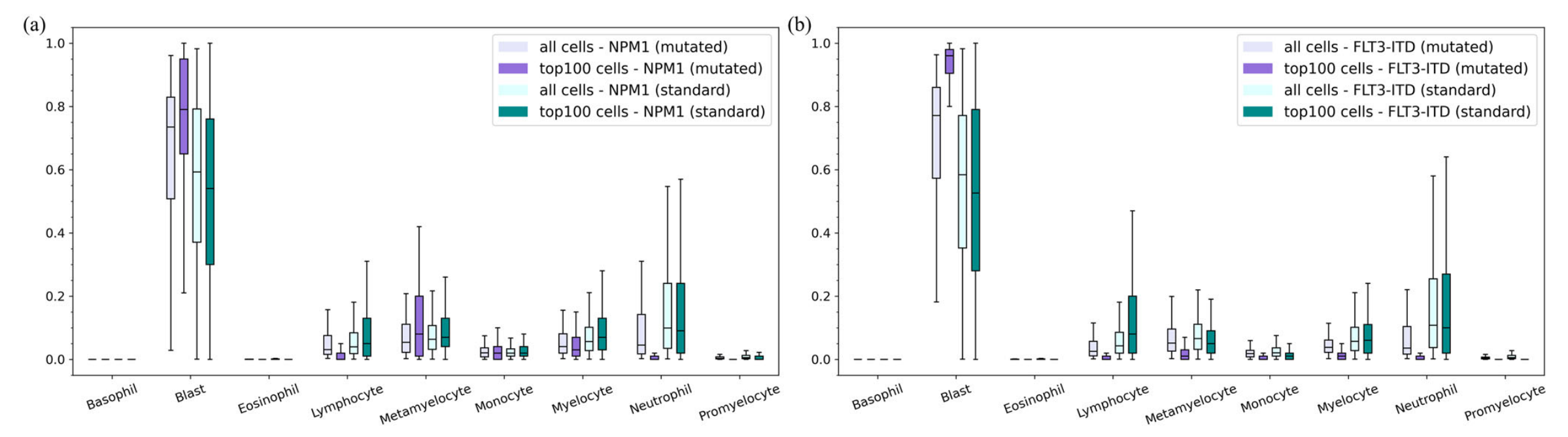
图a(NPM1突变数据)
- 在MIL预测前,计算每个袋中各类细胞的比例(即“all cells - NPM1 (mutated)”和“all cells - NPM1 (standard)” ,分别代表NPM1突变和标准袋中的所有细胞比例) 。
- 预测后,选取每个袋中突变概率最高的前100个细胞(即“top100 cells - NPM1 (mutated)”和“top100 cells - NPM1 (standard)” ,分别对应NPM1突变和标准袋中的前100个细胞比例) ,计算其比例并按细胞类型绘制分布。
- 对于突变袋(紫色相关),原始细胞(Blast)在MIL预测后的前100个细胞中比例显著增加,而其他细胞类型比例相对下降;
- 对于标准袋(青绿色相关),原始细胞在MIL预测后的前100个细胞中比例明显降低,其他细胞类型比例有不同程度上升。
- 这表明原始细胞在NPM1突变预测中具有重要指示作用。
图b(FLT3 - ITD数据)
- 先计算MIL预测前每个袋中各类细胞比例(“all cells - FLT3-ITD (mutated)”和“all cells - FLT3-ITD (standard)” ,分别为FLT3 - ITD突变和标准袋中的所有细胞比例) 。
- 预测后,对每个袋中突变概率最高的前100个细胞(“top100 cells - FLT3-ITD (mutated)”和“top100 cells - FLT3-ITD (standard)” ,分别对应FLT3 - ITD突变和标准袋中的前100个细胞比例) 进行比例计算和按细胞类型绘制分布。
- 结果显示,对于突变袋(紫色相关),原始细胞在MIL预测后的前100个细胞中占比大幅升高,其他细胞类型比例相应减少;
- 对于标准袋(青绿色相关),原始细胞在MIL预测后的前100个细胞中比例明显下降,其他细胞类型比例有所上升。这说明原始细胞在FLT3 - ITD突变预测中也是关键指示细胞类型。
总体而言,这两张图共同表明原始细胞(Blast)在MIL模型预测基因突变(无论是NPM1突变还是FLT3 - ITD突变)时是重要的指示细胞类型,突变袋中原始细胞比例在预测后增加,标准袋中原始细胞比例在预测后减少,凸显了原始细胞在区分突变和标准状态中的重要性 。
这可揭示即使仅使用玻片级标注,哪些细胞对MIL预测基因突变更有意义。
可以观察到,无论对于NPM1还是FLT3 - ITD模型,原始细胞(blasts)的趋势保持一致。在存在任一种基因突变的袋中,应用MIL后,前100个代表性细胞中blasts的比例显著增加。
另一方面,在无基因突变的袋中,应用MIL后,前100个代表性细胞中blasts的比例下降。这一趋势表明blasts的存在是深度学习中确定是否存在突变的重要指标。
此外,在存在基因突变的袋中,其他细胞类型的比例下降,表明机器学习中用于确定突变存在的大多数特征属于blasts类别。
因此,在无突变的袋中,由于在blasts细胞中找不到这些特征,其他细胞类型的比例增加,而blasts的比例显著下降。
五、项目复现流程
5-1:系统要求与环境配置
- Python版本: 3.9
- 依赖安装:
git clone https://github.com/c4lab/AML-WSI.git # 假设项目仓库地址 pip install -r requirement.txt # 安装依赖包 - 其他工具: 确保已安装Jupyter Notebook以运行集成学习模块。
5-2:使用PyHIST生成图像补丁
- 目的:将WSI分割为小补丁,过滤无组织区域。
- 输入:原始WSI文件(
.svs,.tiff等)。 - 输出:包含有效组织的补丁图像。
单张WSI处理
python pyhist.py \
--content-threshold 0.05 \ # 组织区域阈值(低于此值视为背景)
--output /path/to/output_patches \ # 补丁保存路径
--output-downsample 1 \ # 下采样率(1为原始分辨率)
--save-patches \ # 保存补丁
--save-tilecrossed-image \ # 保存拼接预览图
--info "verbose" \ # 输出详细信息
/path/to/your/WSI.svs # 输入WSI路径
批量处理整个WSI目录
python /run/run_pyHIST.py # 需在脚本内配置输入/输出路径
注意:编辑run_pyHIST.py以指定输入目录和参数。
5-3:ROI检测(基于DenseNet121)
- 目的:筛选出包含肿瘤区域的补丁。
- 输入:PyHIST生成的补丁目录。
- 输出:分类为ROI的补丁及概率文件。
单张WSI的补丁检测
python ROI_detection/main.py \
--predict-mode \ # 预测模式
--report-excel \ # 生成Excel报告
--data-path /path/to/pyhist_output \ # PyHIST补丁路径
--threshold 0.8 \ # ROI置信度阈值
--output-dir /path/to/roi_output \ # 结果保存路径
--down-scale 1 \ # 下采样率(与PyHIST一致)
--batch-size 32 # 批量大小(根据GPU调整)
批量处理所有WSI
python /run/run_ROI_detection.py # 需在脚本内配置路径和参数
5-4:细胞检测(基于预训练模型)
- 目的:在ROI补丁中定位单个细胞。
- 输入:ROI检测输出的补丁图像。
- 输出:细胞坐标及分类结果。
下载预训练权重
从 Zenodo 下载best_weights.pth。
检测单个补丁中的细胞
python cell_detection/cell_detection.py \
--input_patch /path/to/roi_patch.png \ # ROI补丁路径
--model_weights /path/to/best_weights.pth \ # 权重文件路径
--out_dir /path/to/cell_output # 结果保存路径
输出文件
• cell_detection.csv:包含细胞坐标、类型(如肿瘤细胞、淋巴细胞)及置信度。
5-5:MIL训练(多示例学习模型)
目的
基于补丁和细胞特征预测基因突变状态。
输入
• 训练/验证库文件(描述补丁与WSI的对应关系及标签)
• 原始WSI图像路径(用于特征提取)
训练命令
python MIL/MIL_train.py \
--output /path/to/mil_model \ # 模型保存路径
--train_lib /path/to/train_lib.pt \ # 训练库文件(需预先准备)
--val_lib /path/to/val_lib.pt \ # 验证库文件
--slide_path /path/to/wsi_images \ # WSI原始图像目录
--model densenet121 \ # 选择预训练模型(如resnet50)
--lr 0.0001 \ # 学习率
--epochs 50 # 训练轮次
关键参数说明
• train_lib/val_lib: 需通过自定义脚本生成,包含补丁路径、WSI标签及元数据。
• slide_path: 原始WSI路径,用于提取高分辨率特征。
5-6:集成学习
- 目的:结合多个模型预测结果提升性能。
- 输入:多个MIL模型的预测结果。
操作
- 运行Jupyter Notebook
ensemble.ipynb。 - 设置参数:
ensemble_lib = "/path/to/predictions" # 各模型预测结果目录 gene = "FLT3" # 目标基因名称 - 执行全部单元格生成集成结果。
注意事项
-
路径一致性:确保各步骤输入/输出路径匹配。
-
数据准备:
• 训练库/验证库需预先处理,建议参考项目文档生成。 -
硬件配置:
• ROI检测和MIL训练需GPU加速(推荐NVIDIA GPU + CUDA)。 -
模型选择:在MIL训练中尝试不同预训练模型(如ResNet、EfficientNet)以优化效果。
通过以上步骤,可完成从WSI处理到基因突变预测的全流程。如有环境或依赖问题,请检查requirement.txt并确保Python版本匹配。
结束语
本期推文的内容就到这里啦,如果需要获取医学AI领域的最新发展动态,请关注小罗的推送!如需进一步深入研究,获取相关资料,欢迎加入我的知识星球!



















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









